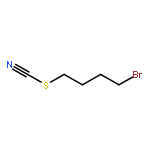
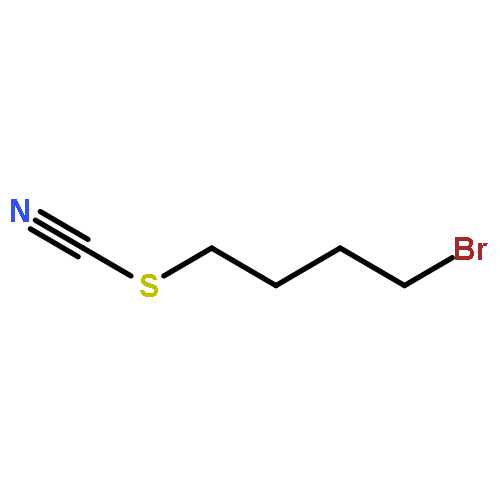
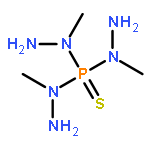
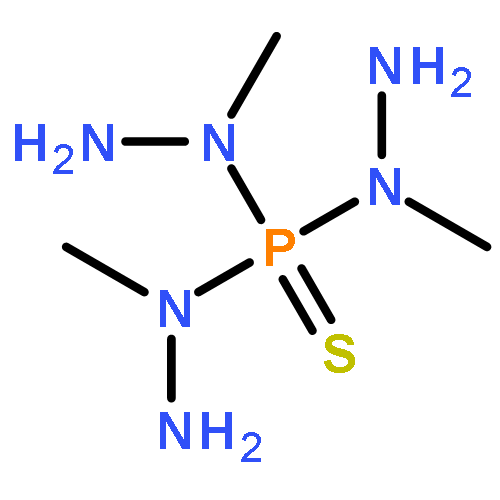
Co-reporter:Nicole Kroll, Kolja Theilacker, Marc Schoknecht, Dirk Baabe, Dennis Wiedemann, Martin Kaupp, Andreas Grohmann and Gerald Hörner
Dalton Transactions 2015 vol. 44(Issue 44) pp:19232-19247
Publication Date(Web):13 Oct 2015
DOI:10.1039/C5DT02502H
The ligand-field strength in metal complexes of polydentate ligands depends critically on how the ligand backbone places the donor atoms in three-dimensional space. Distortions from regular coordination geometries are often observed. In this work, we study the isolated effect of ligand-sphere distortion by means of two structurally related pentadentate ligands of identical donor set, in the solid state (X-ray diffraction, 57Fe-Mössbauer spectroscopy), in solution (NMR spectroscopy, UV/Vis spectroscopy, conductometry), and with quantum-chemical methods. Crystal structures of hexacoordinate iron(II) and nickel(II) complexes derived from the cyclic ligand L1 (6-methyl-6-(pyridin-2-yl)-1,4-bis(pyridin-2-ylmethyl)-1,4-diazepane) and its open-chain congener L2 (N1,N3,2-trimethyl-2-(pyridine-2-yl)-N1,N3-bis(pyridine-2-ylmethyl) propane-1,3-diamine) reveal distinctly different donor set distortions reflecting the differences in ligand topology. Distortion from regular octahedral geometry is minor for complexes of ligand L2, but becomes significant in the complexes of the cyclic ligand L1, where trans elongation of Fe−N bonds cannot be compensated by the rigid ligand backbone. This provokes trigonal twisting of the ligand field. This distortion causes the metal ion in complexes of L1 to experience a significantly weaker ligand field than in the complexes of L2, which are more regular. The reduced ligand-field strength in complexes of L1 translates into a marked preference for the electronic high-spin state, the emergence of conformational isomers, and massively enhanced lability with respect to ligand exchange and oxidation of the central ion. Accordingly, oxoiron(IV) species derived from L1 and L2 differ in their spectroscopic properties and their chemical reactivity.
Co-reporter:Andreas Grohmann;Marco Haryono;Katja Student;Paul Müller;Michael Stocker
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 5-6) pp:662-669
Publication Date(Web):
DOI:10.1002/ejic.201201156
Abstract
This microreview highlights recent techniques used to determine the spin state at or around room temperature in nanoscale objects composed of iron(II) spin-crossover complexes. Complexes have been studied in the form of thin films, nanoparticles, single molecules or oligomolecular aggregates. Three approaches are presented: Surface plasmon polariton resonance in films on Ti/Au substrates, the determination of single-particle current–voltage characteristics between gold electrodes, and current-imaging tunnelling spectroscopy performed on highly oriented pyrolytic graphite-adsorbed strings of oligonuclear, bead-like complex aggregates.
Co-reporter:E. Alper Ünal, Dennis Wiedemann, Josef Seiffert, John P. Boyd, Andreas Grohmann
Tetrahedron Letters 2012 Volume 53(Issue 1) pp:54-55
Publication Date(Web):4 January 2012
DOI:10.1016/j.tetlet.2011.10.131
A variety of substituted tris(pyridyl) frameworks, as well as a pentakis(pyridine) system, are conveniently accessible in a one-pot synthesis, which consists of two sequential nucleophilic aromatic substitutions, employing substituted pyridine precursors.
Co-reporter:Nicole Kroll, Kolja Theilacker, Marc Schoknecht, Dirk Baabe, Dennis Wiedemann, Martin Kaupp, Andreas Grohmann and Gerald Hörner
Dalton Transactions 2015 - vol. 44(Issue 44) pp:NaN19247-19247
Publication Date(Web):2015/10/13
DOI:10.1039/C5DT02502H
The ligand-field strength in metal complexes of polydentate ligands depends critically on how the ligand backbone places the donor atoms in three-dimensional space. Distortions from regular coordination geometries are often observed. In this work, we study the isolated effect of ligand-sphere distortion by means of two structurally related pentadentate ligands of identical donor set, in the solid state (X-ray diffraction, 57Fe-Mössbauer spectroscopy), in solution (NMR spectroscopy, UV/Vis spectroscopy, conductometry), and with quantum-chemical methods. Crystal structures of hexacoordinate iron(II) and nickel(II) complexes derived from the cyclic ligand L1 (6-methyl-6-(pyridin-2-yl)-1,4-bis(pyridin-2-ylmethyl)-1,4-diazepane) and its open-chain congener L2 (N1,N3,2-trimethyl-2-(pyridine-2-yl)-N1,N3-bis(pyridine-2-ylmethyl) propane-1,3-diamine) reveal distinctly different donor set distortions reflecting the differences in ligand topology. Distortion from regular octahedral geometry is minor for complexes of ligand L2, but becomes significant in the complexes of the cyclic ligand L1, where trans elongation of Fe−N bonds cannot be compensated by the rigid ligand backbone. This provokes trigonal twisting of the ligand field. This distortion causes the metal ion in complexes of L1 to experience a significantly weaker ligand field than in the complexes of L2, which are more regular. The reduced ligand-field strength in complexes of L1 translates into a marked preference for the electronic high-spin state, the emergence of conformational isomers, and massively enhanced lability with respect to ligand exchange and oxidation of the central ion. Accordingly, oxoiron(IV) species derived from L1 and L2 differ in their spectroscopic properties and their chemical reactivity.