Co-reporter:Eve M. Mozur, Annalise E. Maughan, Yongqiang Cheng, Ashfia Huq, Niina Jalarvo, Luke L. Daemen, and James R. Neilson
Chemistry of Materials December 12, 2017 Volume 29(Issue 23) pp:10168-10168
Publication Date(Web):November 7, 2017
DOI:10.1021/acs.chemmater.7b04017
Hybrid organic–inorganic perovskites have gained notoriety in the photovoltaic community for their composition-tunable band gaps and long-lived electronic excited states, which are known to be related to the crystalline phase. While it is known that the inorganic and organic components are coupled through structural phase transitions, it remains unclear as to what role each plays in directing the structure of hybrid perovskites such as methylammonium lead halides (CH3NH3PbX3). Here, we present crystallographic and spectroscopic data for the series (CH3NH3)1–xCsxPbBr3. CH3NH3PbBr3 behaves as a plastic crystal in the high temperature cubic phase, and substitution of CH3NH3+ with Cs+ leads to the formation of an orientational glass. While the organic molecule exhibits slow, glassy reorientational dynamics, the inorganic framework continues to undergo crystallographic phase transitions. These crystallographic transitions occur in the absence of thermodynamic signatures in the specific heat from molecular orientation transitions, which suggests that the phase transitions result from underlying instabilities intrinsic to the inorganic lattice. However, these transitions are not decoupled from the reorientations of the organic molecule, as indicated by inelastic and quasielastic neutron scattering. Observation of a reentrant phase transition in (CH3NH3)0.8Cs0.2PbBr3 permits the resolution of these complex behaviors within the context of strain mediated interactions. Together, these results provide critical insight into the coupled phase behavior and dynamics in hybrid perovskites.
Co-reporter:Andrew J. Martinolich and James R. Neilson
Chemistry of Materials 2017 Volume 29(Issue 2) pp:
Publication Date(Web):December 20, 2016
DOI:10.1021/acs.chemmater.6b04861
The control of solid state reaction pathways will enable the design and discovery of new functional inorganic materials. A range of synthetic approaches have been used to shift solid state chemistry away from thermodynamic control, in which the most energetically favorable product forms, toward a regime of kinetic control, so that metastable materials can be controllably produced. In this Perspective, we focus on the kinetic control of solid state metathesis reactions to alter solid state reaction pathways and products. We provide insight into the necessary components of a kinetically controlled solid state reaction and illustrate the utility of studying reactions in situ in order to observe the various intermediates and kinetic pathways that may extend synthetic solid state chemistry toward a paradigm of reaction-by-design.
Co-reporter:Annalise E. Maughan; Alex M. Ganose; Mitchell M. Bordelon; Elisa M. Miller; David O. Scanlon
Journal of the American Chemical Society 2016 Volume 138(Issue 27) pp:8453-8464
Publication Date(Web):June 10, 2016
DOI:10.1021/jacs.6b03207
Vacancy-ordered double perovskites of the general formula A2BX6 are a family of perovskite derivatives composed of a face-centered lattice of nearly isolated [BX6] units with A-site cations occupying the cuboctahedral voids. Despite the presence of isolated octahedral units, the close-packed iodide lattice provides significant electronic dispersion, such that Cs2SnI6 has recently been explored for applications in photovoltaic devices. To elucidate the structure–property relationships of these materials, we have synthesized solid-solution Cs2Sn1–xTexI6. However, even though tellurium substitution increases electronic dispersion via closer I–I contact distances, the substitution experimentally yields insulating behavior from a significant decrease in carrier concentration and mobility. Density functional calculations of native defects in Cs2SnI6 reveal that iodine vacancies exhibit a low enthalpy of formation, and that the defect energy level is a shallow donor to the conduction band rendering the material tolerant to these defect states. The increased covalency of Te–I bonding renders the formation of iodine vacancy states unfavorable and is responsible for the reduction in conductivity upon Te substitution. Additionally, Cs2TeI6 is intolerant to the formation of these defects, because the defect level occurs deep within the band gap and thus localizes potential mobile charge carriers. In these vacancy-ordered double perovskites, the close-packed lattice of iodine provides significant electronic dispersion, while the interaction of the B- and X-site ions dictates the properties as they pertain to electronic structure and defect tolerance. This simplified perspective based on extensive experimental and theoretical analysis provides a platform from which to understand structure–property relationships in functional perovskite halides
Co-reporter:Andrew J. Martinolich, Joshua A. Kurzman, and James R. Neilson
Journal of the American Chemical Society 2016 Volume 138(Issue 34) pp:11031-11037
Publication Date(Web):August 4, 2016
DOI:10.1021/jacs.6b06367
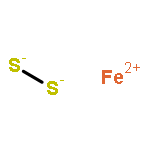
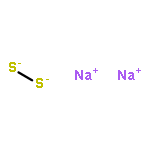
Solid-state diffusion is often the primary limitation in the synthesis of crystalline inorganic materials and prevents the potential discovery and isolation of new materials that may not be the most stable with respect to the reaction conditions. Synthetic approaches that circumvent diffusion in solid-state reactions are rare and often allow the formation of metastable products. To this end, we present an in situ study of the solid-state metathesis reactions MCl2 + Na2S2 → MS2 + 2 NaCl (M = Fe, Co, Ni) using synchrotron powder X-ray diffraction and differential scanning calorimetry. Depending on the preparation method of the reaction, either combining the reactants in an air-free environment or grinding homogeneously in air before annealing, the barrier to product formation, and therefore reaction pathway, can be altered. In the air-free reactions, the product formation appears to be diffusion limited, with a number of intermediate phases observed before formation of the MS2 product. However, grinding the reactants in air allows NaCl to form directly without annealing and displaces the corresponding metal and sulfide ions into an amorphous matrix, as confirmed by pair distribution function analysis. Heating this mixture yields direct nucleation of the MS2 phase and avoids all crystalline binary intermediates. Grinding in air also dissipates a large amount of lattice energy via the formation of NaCl, and the crystallization of the metal sulfide is a much less exothermic process. This approach has the potential to allow formation of a range of binary, ternary, or higher-ordered compounds to be synthesized in the bulk, while avoiding the formation of many binary intermediates that may otherwise form in a diffusion-limited reaction.
Co-reporter:Andrew J. Martinolich, Robert F. Higgins, Matthew P. Shores, and James R. Neilson
Chemistry of Materials 2016 Volume 28(Issue 6) pp:1854
Publication Date(Web):February 19, 2016
DOI:10.1021/acs.chemmater.6b00027
Preparation of metastable solid-state materials is often hindered by the limitations of traditional solid-state chemistry, namely the high temperatures required to induce solid-state diffusion. Here, we present the formation of the metastable pyrite polymorph of CuSe2 through a metathesis reaction mediated by the Lewis base, triphenylphosphine (Ph3P). The Ph3P added to the double salt-exchange reaction CuCl2 + Na2Se2 abstracts selenium from the precursor and promotes the reaction at 130–150 °C, rather than 300 °C required for the neat reaction mixture. Powder X-ray diffraction experiments indicate that substoichiometric amounts of added phosphine yield pyrite CuSe2; infrared and nuclear magnetic resonance spectroscopies as well as mass spectrometry corroborate the formation of Ph3PSe and Cu(Ph3P)n+ adducts, which implicates the Lewis base in facilitating atom-transfer reactions, thus lowering the activation barrier to forming the metastable pyrite polymorph of CuSe2. These results provide an example of a new chemical approach for materials synthesis that will enable the synthesis of new, metastable crystalline materials.
Co-reporter:Mary E. Marisa, Shiliang Zhou, Brent C. Melot, Graham F. Peaslee, and James R. Neilson
Inorganic Chemistry 2016 Volume 55(Issue 23) pp:12290-12298
Publication Date(Web):November 11, 2016
DOI:10.1021/acs.inorgchem.6b02025
Hydroxyapatite is an inorganic mineral closely resembling the mineral phase in bone. However, as a biological mineral, it is highly disordered, and its composition and atomistic structure remain poorly understood. Here, synchrotron X-ray total scattering and pair distribution function analysis methods provide insight into the nature of atomistic disorder in a synthetic bone mineral analogue, chemically substituted hydroxyapatite. By varying the effective hydrolysis rate and/or carbonate concentration during growth of the mineral, compounds with varied degrees of paracrystallinity are prepared. From advanced simulations constrained by the experimental pair distribution function and density functional theory, the paracrystalline disorder prevalent in these materials appears to result from accommodation of carbonate in the lattice through random displacement of the phosphate groups. Though many substitution modalities are likely to occur in concert, the most predominant substitution places carbonate into the mirror plane of an ideal phosphate site. Understanding the mineralogical imperfections of a biologically analogous hydroxyapatite is important not only to potential bone grafting applications but also to biological mineralization processes themselves.
Co-reporter:Andrew J. Martinolich; Joshua A. Kurzman
Journal of the American Chemical Society 2015 Volume 137(Issue 11) pp:3827-3833
Publication Date(Web):March 6, 2015
DOI:10.1021/ja512520z
Rational preparation of materials by design is a major goal of inorganic, solid-state, and materials chemists alike. Oftentimes, the use of nonmetallurgical reactions (e.g., chalcogenide fluxes, hydrothermal syntheses, and in this case solid-state metathesis) alters the thermodynamic driving force of the reaction and allows new, refractory, or otherwise energetically unfavorable materials to form under softer conditions. Taking this a step further, alteration of a metathesis reaction pathway can result in either the formation of the equilibrium marcasite polymorph (by stringent exclusion of air) or the kinetically controlled formation of the high-pressure pyrite polymorph of CuSe2 (by exposure to air). From analysis of the reaction coordinate with in situ synchrotron X-ray diffraction and pair distribution function analysis as well as differential scanning calorimetry, it is clear that the air-exposed reaction proceeds via slight, endothermic rearrangements of crystalline intermediates to form pyrite, which is attributed to partial solvation of the reaction from atmospheric humidity. In contrast, the air-free reaction proceeds via a significant exothermic process to form marcasite. Decoupling the formation of NaCl from the formation of CuSe2 enables kinetic control to be exercised over the resulting polymorph of these superconducting metal dichalcogenides.
Co-reporter:Joshua A. Kurzman, Kevan E. Dettelbach, Andrew J. Martinolich, Curtis P. Berlinguette, and James R. Neilson
Chemistry of Materials 2015 Volume 27(Issue 9) pp:3462
Publication Date(Web):April 13, 2015
DOI:10.1021/acs.chemmater.5b00878
A central challenge for hydrogen generation via electrolytic water splitting is the identification of efficient oxygen evolution reaction (OER) catalysts; a key aspect of the challenge hinges on an ability to relate atomic-scale structure to observed activities. Amorphous iron-based oxide(hydroxides) prepared by photochemical metal organic decomposition (PMOD) are proven OER catalysts, but their atomistic structures have been elusive. Here, a combination of powder diffraction and pair distribution function (PDF) analyses enables the formulation of a set of structural characteristics that capture the salient features of amorphous iron oxide(hydroxide) (a-FeOx), a model compound for this class of materials. a-FeOx contains only octahedrally coordinated iron atoms, which form clusters of both edge- and corner-sharing octahedra. A degree of “eutaxy” with predominantly ABC-type anion stacking persists at length scales beyond the dimensions of cluster domains–consistent with thermally induced crystallization into the defect spinel γ-Fe2O3. Evidence for considerable octahedral irregularities suggests the presence of a large number of bridging and terminal hydroxyl or water ligands, which would provide a high concentration of potential active sites. The structural features of a-FeOx are reminiscent of other first-row transition metal oxyhydroxide OER catalysts that comprise layers of edge-sharing octahedral ions capable of electron transfer and ligand association/dissociation. In keeping with ABC anion stacking, however, the title compound more closely resembles a highly defective spinel lattice rather than a layered hydroxide.
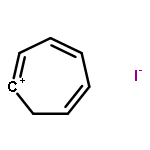
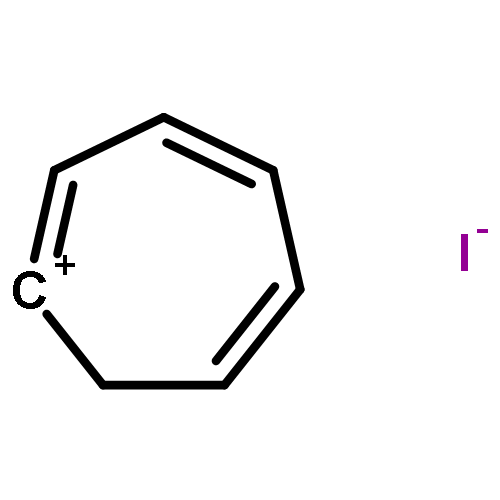
Co-reporter:Annalise E. Maughan; Joshua A. Kurzman
Inorganic Chemistry 2015 Volume 54(Issue 1) pp:370-378
Publication Date(Web):December 19, 2014
DOI:10.1021/ic5025795
Inorganic materials with organic constituents—hybrid materials—have shown incredible promise as chemically tunable functional materials with interesting optical and electronic properties. Here, the preparation and structure are reported of two hybrid materials containing the optoelectronically active tropylium ion within tin- and lead-iodide inorganic frameworks with distinct topologies. The crystal structures of tropylium tin iodide, (C7H7)2SnI6, and tropylium lead iodide, C7H7PbI3, were solved using high-resolution synchrotron powder X-ray diffraction informed by X-ray pair distribution function data and high-resolution time-of-flight neutron diffraction. Tropylium tin iodide contains isolated tin(IV)-iodide octahedra and crystallizes as a deep black solid, while tropylium lead iodide presents one-dimensional chains of face-sharing lead(II)-iodide octahedra and crystallizes as a bright red-orange powder. Experimental diffuse reflectance spectra are in good agreement with density functional calculations of the electronic structure. Calculations of the band decomposed charge densities suggest that the deep black color of tropylium tin iodide is attributed to iodide ligand to tin metal charge transfer, while the bright red-orange color of tropylium lead iodide arises from charge transfer between iodine and tropylium states. Understanding the origins of the observed optoelectronic properties of these two compounds, with respect to their distinct topologies and organic–inorganic interactions, provides insight into the design of tropylium-containing compounds for potential optical and electronic applications.
Co-reporter: James R. Neilson;Dr. Nathan C. George;Dr. Meredith M. Murr; Ram Seshadri; Daniel E. Morse
Chemistry - A European Journal 2014 Volume 20( Issue 17) pp:4956-4965
Publication Date(Web):
DOI:10.1002/chem.201304704
Abstract
Organisms of the phylum Porifera, that is, sponges, utilize enzymatic hydrolysis to concatenate bioavailable inorganic silicon to produce lightweight, strong, and often flexible skeletal elements called spicules. In their optical transparency, these remarkable biomaterials resemble fused silica, despite having been formed under ambient marine biological conditions. Although previous studies have elucidated the chemical mechanisms of spicule formation and revealed the extensive hydration of these glasses, their precise composition and local and medium-range structures had not been determined. We have employed a combination of compositional analysis, 1H and 29Si solid-state nuclear magnetic resonance spectroscopy, and synchrotron X-ray total scattering to characterize spicule-derived silica produced by the demosponge Tethya aurantia. These studies indicate that the materials are highly hydrated, but in an inhomogeneous manner. The spicule-derived silica is, on average, perfectly dense for the given extent of hydration and regions of fully condensed and unstrained SiO networks persist throughout each monolithic spicule. To accommodate chemical strain and defects, the extensive hydration is concentrated in distinct regions that give rise to mesostructural features. The chemistry responsible for producing spicule silica resembles hydrolytic sol-gel processing, which offers exceptional control over the precise local atomic arrangement of materials. However, the specific processing involved in forming the sponge spicule silica further results in regions of fully condensed silica coexisting with regions of incomplete condensation. This mesostructure suggests a mechanism for atomistic defect tolerance and strain relief that may account for the unusual mechanical properties of the biogenic spicules.
Co-reporter:Andrew J. Martinolich
Journal of the American Chemical Society () pp:
Publication Date(Web):October 14, 2014
DOI:10.1021/ja5081647
The preparation of materials with limited phase stabilities yet high kinetic activation barriers is challenging. Knowledge of their possible formation pathways aids in addressing these challenges. Metathesis reactions present an approach to circumvent these barriers; however, solid-state metathesis reactions are often too rapid from extensive self-heating to understand the reaction. The stoichiometric reaction of MCl2 salts (M = Mn, Fe, Co, Ni, Cu, Zn) with Na2S2 enables the formation of pyrite (FeS2), CoS2, and NiS2 at low temperatures (250–350 °C). Na2S2 has the same polyanionic dimer as found in the pyrite structure, which would suggest the possibility of a facile ion-exchange reaction. However, from high-resolution synchrotron X-ray diffraction and differential scanning calorimetry, the energetic driving force does not appear to result solely from NaCl formation but also from formation of intermediate and pyrite phases. It is apparent that the reaction proceeds through polyanionic disproportionation and formation of a low-density alkali-rich intermediate, followed by anionic comproportionation and atomic rearrangement into the pyrite phase. These results have profound implications for the use of low-temperature metathesis in achieving materials by design.
.jpg)