Co-reporter:Zhen Luo, Yingjin Ma, Chungen Liu, and Haibo Ma
Journal of Chemical Theory and Computation October 10, 2017 Volume 13(Issue 10) pp:4699-4699
Publication Date(Web):September 12, 2017
DOI:10.1021/acs.jctc.7b00439
We improve the methodology to construct a complete active space-configuration interaction (CAS-CI) expansion for density-matrix renormalization group (DMRG) wave functions using a matrix-product state representation, inspired by the sampling-reconstructed CAS [SR-CAS; Boguslawski, K.; J. Chem. Phys. 2011, 134, 224101] algorithm. In our scheme, the genetic algorithm, in which the “crossover” and “mutation” processes can be optimized based on quantum information theory, is employed when reconstructing a CAS-CI-type wave function in the Hilbert space. Analysis of results for ground and excited state wave functions of conjugated molecules, transition metal compounds, and a lanthanide complex illustrate that our scheme is very efficient for searching the most important CI expansions in large active spaces.
Co-reporter:Zexing Qu, Chen Yang, and Chungen Liu
The Journal of Physical Chemistry A 2015 Volume 119(Issue 3) pp:442-451
Publication Date(Web):December 23, 2014
DOI:10.1021/jp503220q
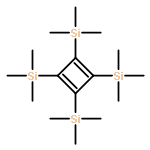
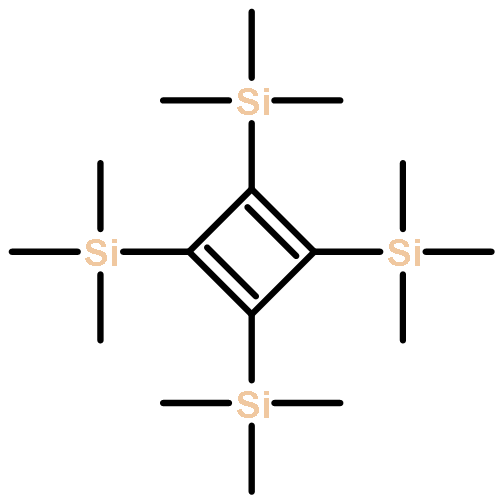
Photoinduced chemical processes upon Franck–Condon (FC) excitation in tetrakis(trimethylsilyl)-cyclobutadiene (TMS-CBD) have been investigated through the exploration of potential energy surface crossings among several low-lying excited states using the complete active space self-consistent field (CASSCF) method. Vertical excitation energies are also computed with the equation-of-motion coupled-cluster model with single and double excitations (EOM-CCSD) as well as the multireference Møller–Plesset (MRMP) methods. Upon finding an excellent coincidence between the computational results and experimental observations, it is suggested that the Franck–Condon excited state does not correspond to the first π–π* single excitation state (S1, 11B1 state in terms of D2 symmetry), but to the second 1B1 state (S3), which is characterized as a σ–π* single excitation state. Starting from the Franck–Condon region, a series of conical intersections (CIs) are located along one isomerization channel and one dissociation channel. Through the isomerization channel, TMS-CBD is transformed to tetrakis(trimethylsilyl)-tetrahedrane (TMS-THD), and this isomerization process could take place by passing through a “tetra form” conical intersection. On the other hand, the dissociation channel yielding two bis(trimethylsilyl)-acetylene (TMS-Ac) molecules through further stretching of the longer C–C bonds might be more competitive than the isomerization channel after excitation into S3 state. This mechanistic picture is in good agreement with recently reported experimental observations.
Co-reporter:Shushu Zhang, Zexing Qu, Peng Tao, Bernard Brooks, Yihan Shao, Xiaoyuan Chen, and Chungen Liu
The Journal of Physical Chemistry C 2012 Volume 116(Issue 23) pp:12434-12442
Publication Date(Web):May 21, 2012
DOI:10.1021/jp3027447
Recent experimental investigation (Reitzenstein and Lambert, Macromolecules2009, 42, 773) indicated that the quite different optical properties of 2,7- and 3,6-linkage triarylboryl carbazole oligomers may arise from the different nature of their low-lying excited states: a low-lying delocalized within-backbone excitation in longer 2,7-linked oligomers vs a backbone-to-side chain charge-transfer (CT) excitation independent of the polymerization length in 3,6-linked oligomers. In this paper, two long-range corrected functionals, CAM-B3LYP and ωB97X, are applied together with the traditional B3LYP functional in time-dependent density functional theory (TDDFT) calculations to systematically investigate thelow-lying electronic excitations in both oligomers. Our calculations indicate that an extensive conjugation exists between monomer molecular orbitals in 2,7-linked oligomers, which is absent in those of 3,6-linked structures, resulting in a considerable narrowing of the HOMO–LUMO gap of their backbone moiety, while having little effect on the side chains. CAM-B3LYP and ωB97x calculations confirm that the lowest-energy absorption is a within-backbone excitation in longer 2,7-linked oligomers as opposed to a backbone to side-chain charge transfer excitation in 2,7-linked oligmers of shorter length and 3,6-linked oligomers of any length. All these findings are consistent with the experimental findings and the qualitative energy diagram proposed by Reitzenstein and Lambert.
Co-reporter:Zexing Qu, Dawei Zhang, Chungen Liu and Yuansheng Jiang
The Journal of Physical Chemistry A 2009 Volume 113(Issue 27) pp:7909-7914
Publication Date(Web):June 15, 2009
DOI:10.1021/jp9015728
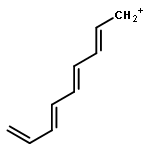
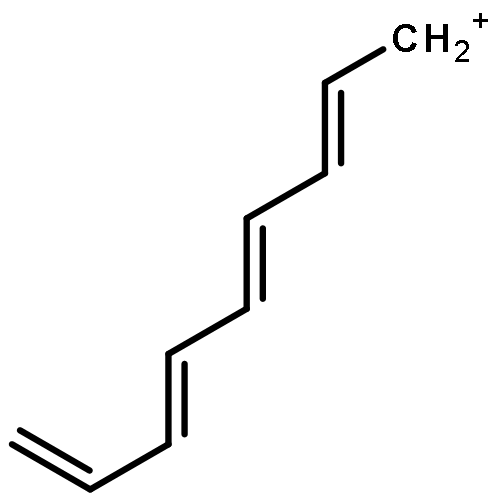
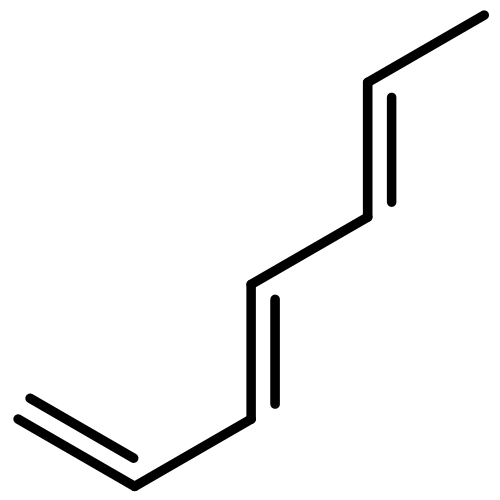
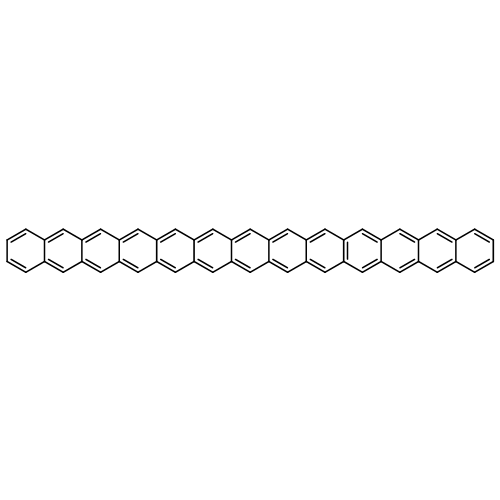
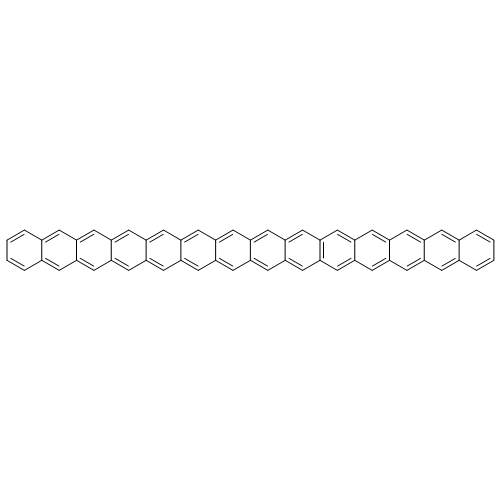
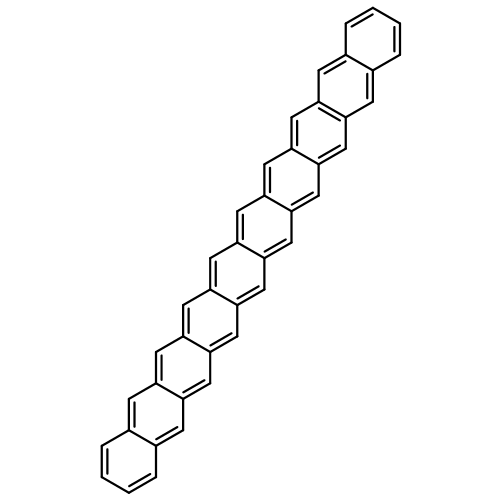
Applying the density matrix renormalization group (DRMG) method to a nonempirical valence bond (VB) model Hamiltonian, we studied polyacene oligomers of different lengths in the strong electron correlation limit. Geometrical optimizations were performed for the lowest singlet and triplet states of oligomers up to [40]-acene, and a convergence of the bond lengths toward the polymer limit is observed in the interior of the oligomer. For large oligomers, as well as for the polymer, the ground state can be reasonably determined to be a singlet. Furthermore, a high similarity between the singlet geometries and triplet geometries suggests an open-shell character for the singlet ground state. A reasonable speculation of the soliton−antisoliton pair character of the singlet ground state was supported by a spin distribution analysis of the triplet state wave function of large oligomers, with each of the two solitons being broadly delocalized over the upper or bottom edge of the oligomers, respectively.
Co-reporter:Hua Lu, ShuShu Zhang, HanZhuang Liu, YanWei Wang, Zhen Shen, ChunGen Liu and XiaoZeng You
The Journal of Physical Chemistry A 2009 Volume 113(Issue 51) pp:14081-14086
Publication Date(Web):December 1, 2009
DOI:10.1021/jp907331q
A boron-dipyrromethene (BODIPY)-based fluorescence probe with a N,N′-(pyridine-2, 6-diylbis(methylene))-dianiline substituent (1) has been prepared by condensation of 2,6-pyridinedicarboxaldehyde with 8-(4-amino)-4,4-difluoro-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene and reduction by NaBH4. The sensing properties of compound 1 toward various metal ions are investigated via fluorometric titration in methanol, which show highly selective fluorescent turn-on response in the presence of Hg2+ over the other metal ions, such as Li+, Na+, K+, Ca2+, Mg2+, Pb2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Cd2+, Ag+, and Mn2+. Computational approach has been carried out to investigate the mechanism why compound 1 provides different fluorescent signal for Hg2+ and other ions. Theoretic calculations of the energy levels show that the quenching of the bright green fluorescence of boradiazaindacene fluorophore is due to the reductive photoinduced electron transfer (PET) from the aniline subunit to the excited state of BODIPY fluorophore. In metal complexes, the frontier molecular orbital energy levels changes greatly. Binding Zn2+ or Cd2+ ion leads to significant decreasing of both the HOMO and LUMO energy levels of the receptor, thus inhibit the reductive PET process, whereas an oxidative PET from the excited state fluorophore to the receptor occurs, vice versa, which also quenches the fluorescence. However, for 1-Hg2+ complex, both the reductive and oxidative PETs are prohibited; therefore, strong fluorescence emission from the fluorophore can be observed experimentally. The agreement of the experimental results and theoretic calculations suggests that our calculation method can be applicable as guidance for the design of new chemosensors for other metal ions.

![Tetracyclo[13.3.1.13,7.19,13]heneicosa-1(19),3,5,7(21),9,11,13(20),15,1
7-nonaene-2,8,14-triyl](http://img.cochemist.com/ccimg/193000/192933-21-4.png)
![Tetracyclo[13.3.1.13,7.19,13]heneicosa-1(19),3,5,7(21),9,11,13(20),15,1
7-nonaene-2,8,14-triyl](http://img.cochemist.com/ccimg/193000/192933-21-4_b.png)


![Silane, tricyclo[1.1.0.02,4]butane-1,2,3,4-tetrayltetrakis[trimethyl-](http://img.cochemist.com/ccimg/352600/352521-08-5.png)
![Silane, tricyclo[1.1.0.02,4]butane-1,2,3,4-tetrayltetrakis[trimethyl-](http://img.cochemist.com/ccimg/352600/352521-08-5_b.png)
![Methyl, [1,1':4',1''-terphenyl]-2,3',3''-triyltris-](http://img.cochemist.com/ccimg/193000/192933-25-8.png)
![Methyl, [1,1':4',1''-terphenyl]-2,3',3''-triyltris-](http://img.cochemist.com/ccimg/193000/192933-25-8_b.png)
![Methyl, [1,1':4',1''-terphenyl]-3,4''-diylbis-](http://img.cochemist.com/ccimg/193000/192933-13-4.png)
![Methyl, [1,1':4',1''-terphenyl]-3,4''-diylbis-](http://img.cochemist.com/ccimg/193000/192933-13-4_b.png)


![Methyl, bis[3-(diphenylmethylene)phenyl]-](http://img.cochemist.com/ccimg/190600/190508-17-9.png)
![Methyl, bis[3-(diphenylmethylene)phenyl]-](http://img.cochemist.com/ccimg/190600/190508-17-9_b.png)