Co-reporter:Nicholas E. S. Tay and David A. Nicewicz
Journal of the American Chemical Society November 15, 2017 Volume 139(Issue 45) pp:16100-16100
Publication Date(Web):October 25, 2017
DOI:10.1021/jacs.7b10076
Nucleophilic aromatic substitution (SNAr) is a direct method for arene functionalization; however, it can be hampered by low reactivity of arene substrates and their availability. Herein we describe a cation radical-accelerated nucleophilic aromatic substitution using methoxy- and benzyloxy-groups as nucleofuges. In particular, lignin-derived aromatics containing guaiacol and veratrole motifs were competent substrates for functionalization. We also demonstrate an example of site-selective substitutive oxygenation with trifluoroethanol to afford the desired trifluoromethylaryl ether.
Co-reporter:Joshua B. McManus and David A. Nicewicz
Journal of the American Chemical Society March 1, 2017 Volume 139(Issue 8) pp:2880-2880
Publication Date(Web):February 8, 2017
DOI:10.1021/jacs.6b12708
Methods for the direct C–H functionalization of aromatic compounds are in demand for a variety of applications, including the synthesis of agrochemicals, pharmaceuticals, and materials. Herein, we disclose the construction of aromatic nitriles via direct C–H functionalization using an acridinium photoredox catalyst and trimethylsilyl cyanide under an aerobic atmosphere. The reaction proceeds at room temperature under mild conditions and has proven to be compatible with a variety of electron-donating and -withdrawing groups, halogens, and nitrogen- and oxygen-containing heterocycles, as well as aromatic-containing pharmaceutical agents.
Co-reporter:Kaila A. Margrey, Joshua B. McManus, Simone Bonazzi, Frederic Zecri, and David A. Nicewicz
Journal of the American Chemical Society August 16, 2017 Volume 139(Issue 32) pp:11288-11288
Publication Date(Web):July 18, 2017
DOI:10.1021/jacs.7b06715
Direct C–H functionalization of aromatic compounds is a useful synthetic strategy that has garnered much attention because of its application to pharmaceuticals, agrochemicals, and late-stage functionalization reactions on complex molecules. On the basis of previous methods disclosed by our lab, we sought to develop a predictive model for site selectivity and extend this aryl functionalization chemistry to a selected set of heteroaromatic systems commonly used in the pharmaceutical industry. Using electron density calculations, we were able to predict the site selectivity of direct C–H functionalization in a number of heterocycles and identify general trends observed across heterocycle classes.
Co-reporter:Leifeng Wang;Fengjin Wu;Dr. Jiean Chen; Dr. David A. Nicewicz; Dr. Yong Huang
Angewandte Chemie International Edition 2017 Volume 56(Issue 24) pp:6896-6900
Publication Date(Web):2017/06/06
DOI:10.1002/anie.201702940
AbstractWe report a formal [4+2] cycloaddition reaction of styrenes under visible-light catalysis. Two styrene molecules with different electronic or steric properties were found to react with each other in good yield and excellent chemo- and regioselectivity. This reaction provides direct access to polysubstituted tetralin scaffolds from readily available styrenes. Sophisticated tricyclic and tetracyclic tetralin analogues were prepared in high yield and up to 20/1 diasteroselectivity from cyclic substrates.
Co-reporter:Kaila A. Margrey;Dr. Alison Levens; David A. Nicewicz
Angewandte Chemie International Edition 2017 Volume 56(Issue 49) pp:15644-15648
Publication Date(Web):2017/12/04
DOI:10.1002/anie.201709523
AbstractThe direct catalytic C−H amination of arenes is a powerful synthetic strategy with useful applications in pharmaceuticals, agrochemicals, and materials chemistry. Despite the advances in catalytic C−H functionalization, the use of aliphatic amine coupling partners is limited. Described herein is the construction of C−N bonds, using primary amines, by direct C−H functionalization with an acridinium photoredox catalyst under an aerobic atmosphere. A wide variety of primary amines, including amino acids and more complex amines are competent coupling partners. Various electron-rich aromatics and heteroaromatics are useful scaffolds in this reaction, as are complex, biologically active arenes. We also describe the ability to functionalize arenes that are not oxidized by an acridinium catalyst, such as benzene and toluene, thus supporting a reactive amine cation radical intermediate.
Co-reporter:Kaila A. Margrey;Dr. Alison Levens; David A. Nicewicz
Angewandte Chemie 2017 Volume 129(Issue 49) pp:15850-15854
Publication Date(Web):2017/12/04
DOI:10.1002/ange.201709523
AbstractThe direct catalytic C−H amination of arenes is a powerful synthetic strategy with useful applications in pharmaceuticals, agrochemicals, and materials chemistry. Despite the advances in catalytic C−H functionalization, the use of aliphatic amine coupling partners is limited. Described herein is the construction of C−N bonds, using primary amines, by direct C−H functionalization with an acridinium photoredox catalyst under an aerobic atmosphere. A wide variety of primary amines, including amino acids and more complex amines are competent coupling partners. Various electron-rich aromatics and heteroaromatics are useful scaffolds in this reaction, as are complex, biologically active arenes. We also describe the ability to functionalize arenes that are not oxidized by an acridinium catalyst, such as benzene and toluene, thus supporting a reactive amine cation radical intermediate.
Co-reporter:Leifeng Wang;Fengjin Wu;Dr. Jiean Chen; Dr. David A. Nicewicz; Dr. Yong Huang
Angewandte Chemie 2017 Volume 129(Issue 24) pp:7000-7004
Publication Date(Web):2017/06/06
DOI:10.1002/ange.201702940
AbstractWe report a formal [4+2] cycloaddition reaction of styrenes under visible-light catalysis. Two styrene molecules with different electronic or steric properties were found to react with each other in good yield and excellent chemo- and regioselectivity. This reaction provides direct access to polysubstituted tetralin scaffolds from readily available styrenes. Sophisticated tricyclic and tetracyclic tetralin analogues were prepared in high yield and up to 20/1 diasteroselectivity from cyclic substrates.
Co-reporter:Kaila A. Margrey and David A. Nicewicz
Accounts of Chemical Research 2016 Volume 49(Issue 9) pp:1997
Publication Date(Web):September 2, 2016
DOI:10.1021/acs.accounts.6b00304
The development of methods for anti-Markovnikov alkene hydrofunctionalization has been a focal point of catalysis research for several decades. The vast majority of work on the control of regioselectivity for this reaction class has hinged on transition metal catalyst activation of olefin substrates. While progress has been realized, there are significant limitations to this approach, and a general solution for catalysis of anti-Markovnikov hydrofunctionalization reactions of olefins does not presently exist.In the past several years, this research lab has focused on alkene activation by single electron oxidation using organic photoredox catalysts to facilitate anti-Markovnikov hydrofunctionalization. By accessing reactive cation radical intermediates, we have realized a truly general approach to anti-Markovnikov olefin hydrofunctionalization reactions. We have identified a dual organic catalyst system consisting of an acridinium photooxidant, first reported by Fukuzumi, and a redox-active hydrogen atom donor that accomplishes a wide range of hydrofunctionalization reactions with complete anti-Markovnikov regiocontrol. This method relies on single electron oxidation of the alkene to reverse its polarity and results in the opposite regioselectivity for hydrofunctionalization.In 2012, we disclosed the anti-Markovnikov hydroetherification of alkenols employing an acridinium photocatalyst and a hydrogen atom donor that proceeds via interwoven polar and radical steps. This general catalyst system has enabled several important reactions in this area, including anti-Markovnikov alkene hydroacetoxylation, hydrolactonization, hydroamination, and hydrotrifluoromethylation reactions. More recently, we have also delineated conditions for intermolecular anti-Markovnikov hydroamination reactions of alkenes using either triflamide or nitrogen-containing heteroaromatic compounds such as pyrazole, indazole, imidazole, and 1,2,3-triazole. Further development led to a method for the anti-Markovnikov addition of mineral acids to olefins using lutidinium halide salts as convenient reagents to deliver the mineral acids. Acids including HCl, HF, H3PO4, and MeSO3H all participate in the hydrofunctionalization reactions, even with alkenes that are highly prone to polymerization.A combination of transient and steady-state absorption spectroscopy tools were employed to observe alkene cation radicals and the resultant acridine radical, lending support for an electron transfer mechanism. The origin of the anti-Markovnikov selectivity in these reactions is likely the result of a reversible addition of the nucleophile to the alkene cation radical resulting in a greater population of the more stable radical. Loss of a proton followed by reaction of the radical intermediate with the hydrogen atom donor completes the transformations. Again, by means of transient absorption spectroscopy, oxidative turnover of the acridine radical was observed to complete the dual catalytic cycle mechanistic picture.
Co-reporter:Andrew J. Perkowski; Wei You
Journal of the American Chemical Society 2015 Volume 137(Issue 24) pp:7580-7583
Publication Date(Web):June 8, 2015
DOI:10.1021/jacs.5b03733
Metal-free, visible light-initiated, living cationic polymerization of 4-methoxystyrene using 2,4,6-tri(p-tolyl)pyrylium tetrafluoroborate and methanol is demonstrated. Molecular weight and dispersity are controlled by the concentration of methanol. Initial mechanistic analysis suggests that methanol likely serves to regulate propagation of the cation chain end via reversible chain transfer in a manner analogous to reversible addition–fragmentation chain transfer polymerization.
Co-reporter:Jeremy D. Griffin; Mary A. Zeller
Journal of the American Chemical Society 2015 Volume 137(Issue 35) pp:11340-11348
Publication Date(Web):August 20, 2015
DOI:10.1021/jacs.5b07770
A direct, catalytic hydrodecarboxylation of primary, secondary, and tertiary carboxylic acids is reported. The catalytic system consists of a Fukuzumi acridinium photooxidant with phenyldisulfide acting as a redox-active cocatalyst. Substoichiometric quantities of Hünig’s base are used to reveal the carboxylate. Use of trifluoroethanol as a solvent allowed for significant improvements in substrate compatibilities, as the method reported is not limited to carboxylic acids bearing α heteroatoms or phenyl substitution. This method has been applied to the direct double decarboxylation of malonic acid derivatives, which allows for the convenient use of dimethyl malonate as a methylene synthon. Kinetic analysis of the reaction is presented showing a lack of a kinetic isotope effect when generating deuterothiophenol in situ as a hydrogen atom donor. Further kinetic analysis demonstrated first-order kinetics with respect to the carboxylate, while the reaction is zero-order in acridinium catalyst, consistent with another finding suggesting the reaction is light limiting and carboxylate oxidation is likely turnover limiting. Stern–Volmer analysis was carried out in order to determine the efficiency for the carboxylates to quench the acridinium excited state.
Co-reporter:Andrew J. Perkowski; Cole L. Cruz
Journal of the American Chemical Society 2015 Volume 137(Issue 50) pp:15684-15687
Publication Date(Web):December 8, 2015
DOI:10.1021/jacs.5b11800
The Newman–Kwart rearrangement is perhaps the quintessential method for the synthesis of thiophenols from the corresponding phenol. However, the high thermal conditions required for the rearrangement of the requisite O-aryl carbamothioates often leads to decomposition. Herein, we present a general strategy for catalysis of O-aryl carbamothioates to S-aryl carbamothioates using catalytic quantities of a commercially available organic single-electron photooxidant. Importantly, this reaction is facilitated at ambient temperatures.
Co-reporter:Peter D. Morse and David A. Nicewicz
Chemical Science 2015 vol. 6(Issue 1) pp:270-274
Publication Date(Web):2014/09/23
DOI:10.1039/C4SC02331E
A direct method to construct 2-oxazolines and 2-thiazolines from corresponding allylic amides and thioamides is reported. The redox-neutral intramolecular hydrofunctionalization is enabled by a dual catalyst system comprised of 9-mesityl-N-methyl acridinium tetrafluoroborate and phenyl disulphide and exhibits complete selectivity for the anti-Markovnikov regioisomeric products. The cyclization of allylic thioamides is postulated to operate via a modified mechanism in which oxidation of the thioamide, rather than the alkene, is responsible for the observed reactivity.
Co-reporter:Nathan J. Gesmundo, Jean-Marc M. Grandjean, and David A. Nicewicz
Organic Letters 2015 Volume 17(Issue 5) pp:1316-1319
Publication Date(Web):February 19, 2015
DOI:10.1021/acs.orglett.5b00316
In this work we present a direct catalytic synthesis of γ-lactams and pyrrolidines from alkenes and activated unsaturated amides or protected unsaturated amines, respectively. Using a mesityl acridinium single electron photooxidant and a thiophenol cocatalyst under irradiation, we are able to directly forge these important classes of heterocycles with complete regiocontrol.
Co-reporter:Cortney L. Cavanaugh and David A. Nicewicz
Organic Letters 2015 Volume 17(Issue 24) pp:6082-6085
Publication Date(Web):December 8, 2015
DOI:10.1021/acs.orglett.5b03113
A direct catalytic synthesis of substituted α-benzyloxyamino-γ-butyrolactones is reported, starting from simple oxime acids and alkenes. The substituted O-benzyloxime acid starting materials are cyclized with oxidizable alkenes, via Polar Radical Crossover Cycloaddition (PRCC) reactions. The catalytic reaction is carried out using the Fukuzumi acridinium photooxidant and substoichiometric amounts of a redox-active cocatalyst. The utility of this method has been demonstrated through the use of 3 oxime acids and 19 oxidizable olefins.
Co-reporter:Nathan A. Romero;Kaila A. Margrey;Nicholas E. Tay
Science 2015 Volume 349(Issue 6254) pp:
Publication Date(Web):
DOI:10.1126/science.aac9895
Lighting the way to aryl C-N bonding
Medicinal chemists like to add N bonds to the C atoms of aromatic rings to make bioactive compounds. By harnessing the energy in visible light, Romero et al. made these links and transformed C-H into C-N bonds. They used a blue-absorbing acridinium ion to activate a ring C for an incoming N partner. A nitroxyl radical co-catalyst (TEMPO) then choreographed the transfer of the H atom to O. The reaction worked for a broad range of substrates, including ammonium as a N source.
Science, this issue p. 1326
Co-reporter:Nathan A. Romero
Journal of the American Chemical Society 2014 Volume 136(Issue 49) pp:17024-17035
Publication Date(Web):November 12, 2014
DOI:10.1021/ja506228u
We describe our efforts to understand the key mechanistic aspects of the previously reported alkene hydrofunctionalization reactions using 9-mesityl-10-methylacridinium (Mes-Acr+) as a photoredox catalyst. Importantly, we are able to detect alkene cation radical intermediates, and confirm that phenylthiyl radical is capable of oxidizing the persistent acridinyl radical in a fast process that unites the catalytic activity of the photoredox and hydrogen atom transfer (HAT) manifolds. Additionally, we present evidence that diphenyl disulfide ((PhS)2) operates on a common catalytic cycle with thiophenol (PhSH) by way of photolytic cleaveage of the disulfide bond. Transition structure analysis of the HAT step using DFT reveals that the activation barrier for H atom donation from PhSH is significantly lower than 2-phenylmalononitrile (PMN) due to structural reorganization. In the early stages of the reaction, Mes-Acr+ is observed to engage in off-cycle adduct formation, presumably as buildup of PhS− becomes significant. The kinetic differences between PhSH and (PhS)2 as HAT catalysts indicate that the proton transfer step may have significant rate limiting influence.
Co-reporter:David A. Nicewicz and Tien M. Nguyen
ACS Catalysis 2014 Volume 4(Issue 1) pp:355
Publication Date(Web):December 11, 2013
DOI:10.1021/cs400956a
Co-reporter:Mary A. Zeller, Michelle Riener, and David A. Nicewicz
Organic Letters 2014 Volume 16(Issue 18) pp:4810-4813
Publication Date(Web):September 5, 2014
DOI:10.1021/ol5022993
A direct catalytic synthesis of γ-butyrolactones from simple alkene and unsaturated acid starting materials is reported. The catalytic system consists of the Fukuzumi acridinium photooxidant and substoichiometric quantities of a redox-active cocatalyst. Oxidizable alkenes such as styrenes and trisubstituted aliphatic alkenes are cyclized with unsaturated acids via polar radical crossover cycloaddition (PRCC) reactions. This method has been applied to the diastereoselective total synthesis of methylenolactocin and protolichesterinic acid.
Co-reporter:Tien M. Nguyen;Namita Manohar ; David A. Nicewicz
Angewandte Chemie International Edition 2014 Volume 53( Issue 24) pp:6198-6201
Publication Date(Web):
DOI:10.1002/anie.201402443
Abstract
Disclosed herein is a general catalytic system for the intermolecular anti-Markovnikov hydroamination of alkenes. By using an organocatalytic photoredox system, α- and β-substituted styrenes as well as aliphatic alkenes undergo anti-Markovnikov hydroamination. Heterocyclic amines were also successfully employed as nitrogen nucleophiles, thus providing a direct route to heterocyclic motifs common in medicinal agents.
Co-reporter:Tien M. Nguyen;Namita Manohar ; David A. Nicewicz
Angewandte Chemie 2014 Volume 126( Issue 24) pp:6312-6315
Publication Date(Web):
DOI:10.1002/ange.201402443
Abstract
Disclosed herein is a general catalytic system for the intermolecular anti-Markovnikov hydroamination of alkenes. By using an organocatalytic photoredox system, α- and β-substituted styrenes as well as aliphatic alkenes undergo anti-Markovnikov hydroamination. Heterocyclic amines were also successfully employed as nitrogen nucleophiles, thus providing a direct route to heterocyclic motifs common in medicinal agents.
Co-reporter:Tien M. Nguyen and David A. Nicewicz
Journal of the American Chemical Society 2013 Volume 135(Issue 26) pp:9588-9591
Publication Date(Web):June 14, 2013
DOI:10.1021/ja4031616
Herein we report a metal-free method for the direct anti-Markovnikov hydroamination of unsaturated amines. Irradiation of the amine substrates with visible light in the presence of catalytic quantities of easily synthesized 9-mesityl-10-methylacridinium tetrafluoroborate and thiophenol as a hydrogen-atom donor furnished the nitrogen-containing heterocycles with complete regiocontrol. Two examples of intermolecular anti-Markovnikov alkene hydroamination are also disclosed.
Co-reporter:Andrew J. Perkowski
Journal of the American Chemical Society 2013 Volume 135(Issue 28) pp:10334-10337
Publication Date(Web):July 1, 2013
DOI:10.1021/ja4057294
A direct catalytic anti-Markovnikov addition of carboxylic acids to alkenes is reported. The catalyst system is comprised of the Fukuzumi acridinium photooxidant (1) and a substoichiometric quantity of a hydrogen-atom donor. Oxidizable olefins, such as styrenes, trisubstituted aliphatic alkenes, and enamides, can be employed along with a variety of carboxylic acids to afford the anti-Markovnikov addition adducts exclusively. A deuterium-labeling experiment lends insight to the potential mechanism.
Co-reporter:Michelle Riener and David A. Nicewicz
Chemical Science 2013 vol. 4(Issue 6) pp:2625-2629
Publication Date(Web):15 Apr 2013
DOI:10.1039/C3SC50643F
A direct method to synthesize lignan cyclobutanes and analogs via photoinduced electron transfer is presented. A variety of oxygenated alkenes are employed to furnish terminal or substituted cyclobutane adducts with complete regiocontrol, yielding cycloadducts with trans stereochemistry. Key to minimizing competing cycloreversion is the inclusion of an aromatic electron relay (ER). This method has been adapted to the synthesis of the natural products magnosalin and pellucidin A.
Co-reporter:Dale J. Wilger, Nathan J. Gesmundo and David A. Nicewicz
Chemical Science 2013 vol. 4(Issue 8) pp:3160-3165
Publication Date(Web):21 May 2013
DOI:10.1039/C3SC51209F
Herein is presented a direct method for the metal-free hydrotrifluoromethylation of alkenes. The method relies on the single electron oxidation of a commercially available sodium trifluoromethanesulfinate salt (CF3SO2Na, Langlois reagent) by N-Me-9-mesityl acridinium as a photoredox catalyst. Methyl thiosalicylate is used as a substoichiometric H-atom donor for aliphatic alkenes, and thiophenol is used as a stoichiometric H-atom donor for styrenyl substrates. The substrate scope for the transformation is broad, including mono-, di- and trisubstituted aliphatic and styrenyl alkenes, with high regioselectivity in nearly all cases examined.
Co-reporter:Jean-Marc M. Grjean ; David A. Nicewicz
Angewandte Chemie 2013 Volume 125( Issue 14) pp:4059-4063
Publication Date(Web):
DOI:10.1002/ange.201210111
Co-reporter:Jean-Marc M. Grjean ; David A. Nicewicz
Angewandte Chemie International Edition 2013 Volume 52( Issue 14) pp:3967-3971
Publication Date(Web):
DOI:10.1002/anie.201210111
Co-reporter:David S. Hamilton
Journal of the American Chemical Society 2012 Volume 134(Issue 45) pp:18577-18580
Publication Date(Web):October 31, 2012
DOI:10.1021/ja309635w
A direct intramolecular anti-Markovnikov hydroetherification reaction of alkenols is described. By employing catalytic quantities of commercially available 9-mesityl-10-methylacridinium perchlorate and 2-phenylmalononitrile as a redox-cycling source of a H-atom, we report the anti-Markovnikov hydroetherification of alkenes with complete regioselectivity. In addition, we present results demonstrating that this novel catalytic system can be applied to the anti-Markovnikov hydrolactonization of alkenoic acids.
Co-reporter:Peter D. Morse and David A. Nicewicz
Chemical Science (2010-Present) 2015 - vol. 6(Issue 1) pp:NaN274-274
Publication Date(Web):2014/09/23
DOI:10.1039/C4SC02331E
A direct method to construct 2-oxazolines and 2-thiazolines from corresponding allylic amides and thioamides is reported. The redox-neutral intramolecular hydrofunctionalization is enabled by a dual catalyst system comprised of 9-mesityl-N-methyl acridinium tetrafluoroborate and phenyl disulphide and exhibits complete selectivity for the anti-Markovnikov regioisomeric products. The cyclization of allylic thioamides is postulated to operate via a modified mechanism in which oxidation of the thioamide, rather than the alkene, is responsible for the observed reactivity.
Co-reporter:Michelle Riener and David A. Nicewicz
Chemical Science (2010-Present) 2013 - vol. 4(Issue 6) pp:NaN2629-2629
Publication Date(Web):2013/04/15
DOI:10.1039/C3SC50643F
A direct method to synthesize lignan cyclobutanes and analogs via photoinduced electron transfer is presented. A variety of oxygenated alkenes are employed to furnish terminal or substituted cyclobutane adducts with complete regiocontrol, yielding cycloadducts with trans stereochemistry. Key to minimizing competing cycloreversion is the inclusion of an aromatic electron relay (ER). This method has been adapted to the synthesis of the natural products magnosalin and pellucidin A.
Co-reporter:Dale J. Wilger, Nathan J. Gesmundo and David A. Nicewicz
Chemical Science (2010-Present) 2013 - vol. 4(Issue 8) pp:NaN3165-3165
Publication Date(Web):2013/05/21
DOI:10.1039/C3SC51209F
Herein is presented a direct method for the metal-free hydrotrifluoromethylation of alkenes. The method relies on the single electron oxidation of a commercially available sodium trifluoromethanesulfinate salt (CF3SO2Na, Langlois reagent) by N-Me-9-mesityl acridinium as a photoredox catalyst. Methyl thiosalicylate is used as a substoichiometric H-atom donor for aliphatic alkenes, and thiophenol is used as a stoichiometric H-atom donor for styrenyl substrates. The substrate scope for the transformation is broad, including mono-, di- and trisubstituted aliphatic and styrenyl alkenes, with high regioselectivity in nearly all cases examined.
.jpg)














![2-Propynoic acid, 3-[tris(1-methylethyl)silyl]-](http://img.cochemist.com/ccimg/839700/839697-82-4.png)
![2-Propynoic acid, 3-[tris(1-methylethyl)silyl]-](http://img.cochemist.com/ccimg/839700/839697-82-4_b.png)













![Propanoic acid, 2-[(phenylmethoxy)imino]-, (2E)-](http://img.cochemist.com/ccimg/189200/189169-23-1.png)
![Propanoic acid, 2-[(phenylmethoxy)imino]-, (2E)-](http://img.cochemist.com/ccimg/189200/189169-23-1_b.png)






![2-Butanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/120600/120591-36-8.png)
![2-Butanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/120600/120591-36-8_b.png)



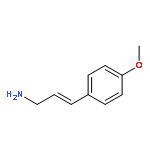
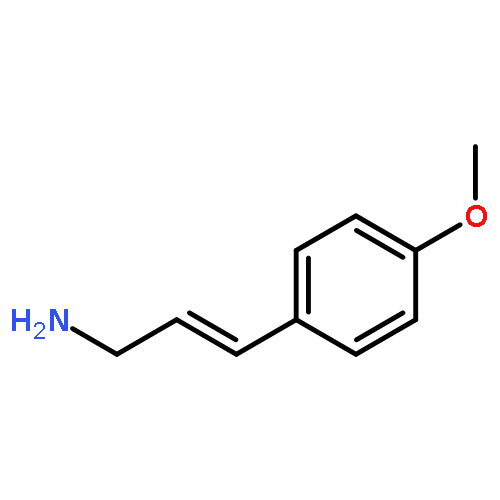










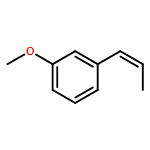
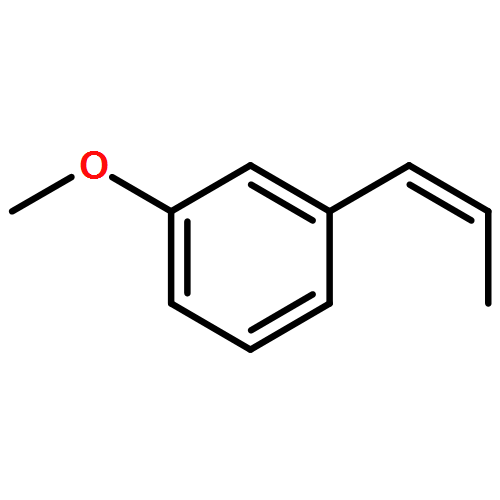








![Phosphonium, [2-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)ethyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/65300/65273-64-5.png)
![Phosphonium, [2-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)ethyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/65300/65273-64-5_b.png)




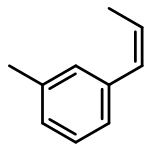
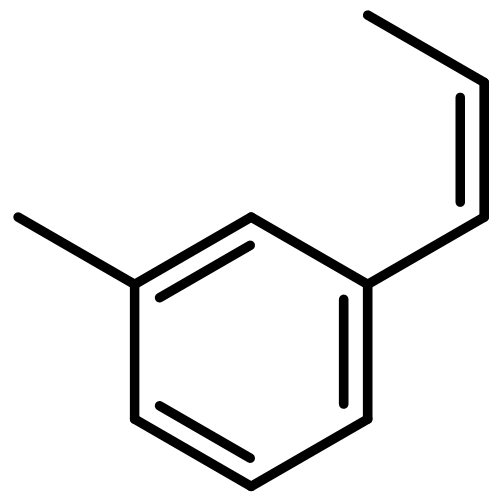
![1-methoxy-3-[(1E)-prop-1-en-1-yl]benzene](http://img.cochemist.com/ccimg/53000/52956-26-0.png)
![1-methoxy-3-[(1E)-prop-1-en-1-yl]benzene](http://img.cochemist.com/ccimg/53000/52956-26-0_b.png)







![Benzeneacetic acid, α-[(phenylmethoxy)imino]-, (Z)- (9CI)](/data/chemimg/1836400/39854-77-8.png)
![Benzeneacetic acid, α-[(phenylmethoxy)imino]-, (Z)- (9CI)](/data/chemimg/1836400/39854-77-8_b.png)




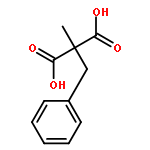
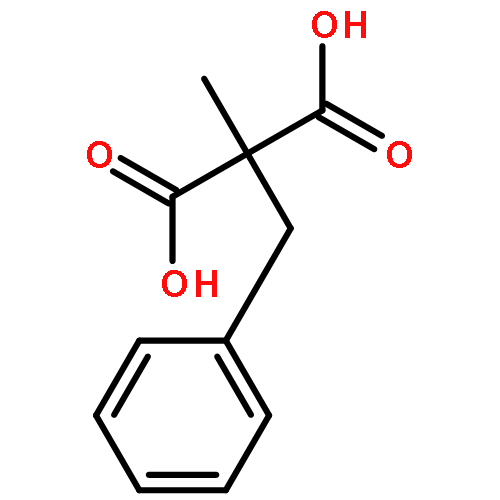

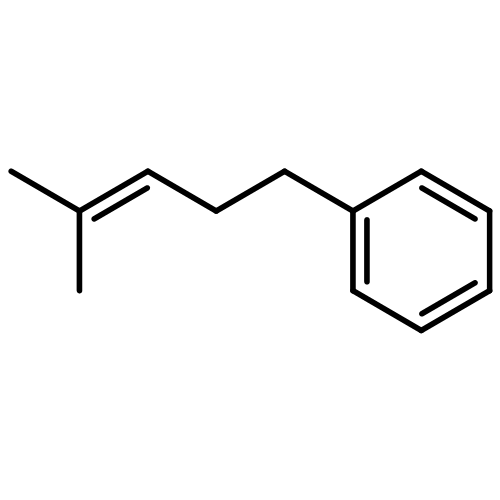


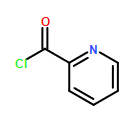



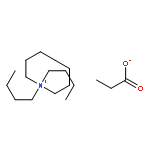
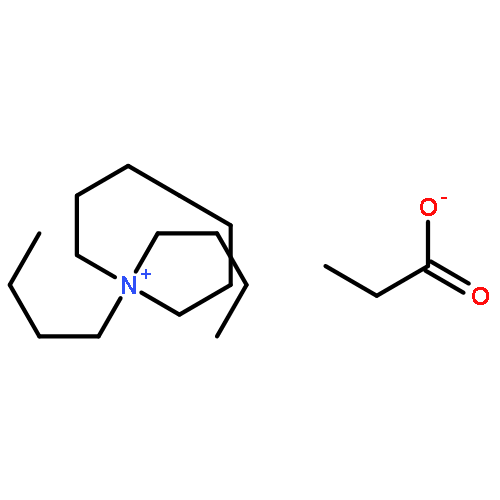








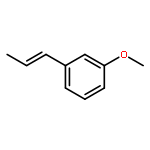



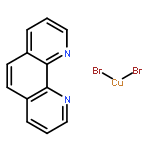
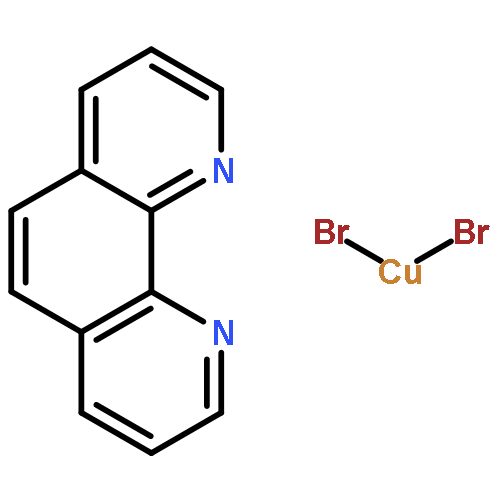
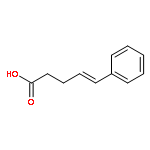
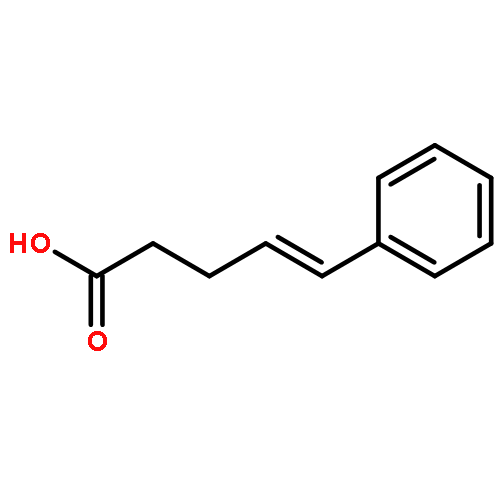


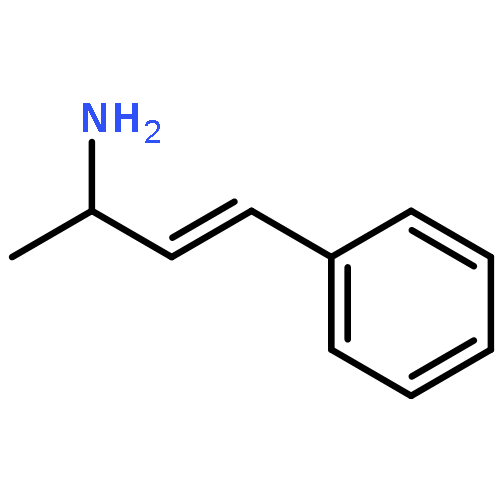
![Carbamothioic acid, dimethyl-, S-[1,1'-biphenyl]-4-yl ester](http://img.cochemist.com/ccimg/16300/16241-09-1.png)
![Carbamothioic acid, dimethyl-, S-[1,1'-biphenyl]-4-yl ester](http://img.cochemist.com/ccimg/16300/16241-09-1_b.png)




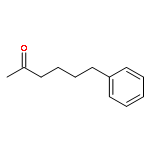
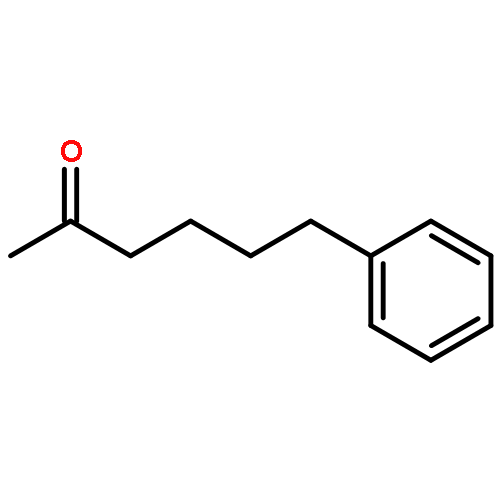
![O-[4-(acetylamino)phenyl] dimethylcarbamothioate](http://img.cochemist.com/ccimg/13600/13522-65-1.png)
![O-[4-(acetylamino)phenyl] dimethylcarbamothioate](http://img.cochemist.com/ccimg/13600/13522-65-1_b.png)


![S-[4-(acetylamino)phenyl] dimethylcarbamothioate](http://img.cochemist.com/ccimg/13600/13512-00-0.png)
![S-[4-(acetylamino)phenyl] dimethylcarbamothioate](http://img.cochemist.com/ccimg/13600/13512-00-0_b.png)


















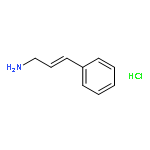

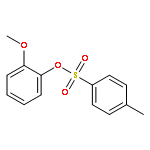

























![4-[(1E)-prop-1-en-1-yl]phenol](http://img.cochemist.com/ccimg/600/539-12-8.png)
![4-[(1E)-prop-1-en-1-yl]phenol](http://img.cochemist.com/ccimg/600/539-12-8_b.png)

