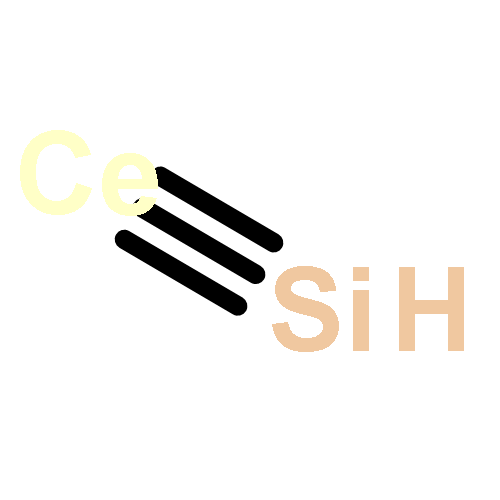
Co-reporter:Shuang He
Journal of Cluster Science 2017 Volume 28( Issue 4) pp:2309-2322
Publication Date(Web):08 May 2017
DOI:10.1007/s10876-017-1225-x
The geometries, electronic structures and properties including simulated photoelectron spectra (PES), adiabatic electron affinities (AEAs), and relative stability of LuSin (n = 3–10) and their anions were investigated adopting the ABCluster global search technique combined with density functional methods. The results revealed that the most stable structures of neutral belong to “substitutional structure”, but not for their anions. The additional electron effects on the most stable structure are intense. The TPSSh AEAs of LuSin (n = 6–9) agree excellently with the experimental data. The mean absolute error and the largest error are only 0.03 eV and 0.05 eV, respectively. The agreement between the experimental and theoretical PES indicates that the most stable structures of LuSin− (n = 6–10) are trustworthy. The DEs and charge transfer are calculated to explain the relative stabilities. HOMO–LUMO gaps reveal that introducing Lu atom to Sin (n = 3–10) raises the photochemical sensitivity.
Co-reporter:Shuang He
Theoretical Chemistry Accounts 2017 Volume 136( Issue 8) pp:93
Publication Date(Web):04 August 2017
DOI:10.1007/s00214-017-2126-7
The equilibrium geometries, electronic structures and electronic properties of PmSin (n = 3–10) clusters were systematically investigated using the ABCluster global search technique combined with density functional methods. The results revealed that the most stable structure of neutral PmSin and their anions can be viewed as replacing a Si atom of the ground state structure of Sin+1 with a Pm atom. The adiabatic electron affinities of PmSin are evaluated, and they differ little from those of SmSin and EuSin. Analyses of HOMO–LUMO gaps showed that introducing Pm atom to Si cluster can significantly improve photochemical reactivity of the cluster. And the improved effects are as good as those of the introducing Sm and Eu atom to Si cluster. The NPA calculations indicated that the 4f electrons of Pm atom in PmSin (n = 3–10) clusters hardly participate in bonding and provide the total magnetic moments. Dissociation energy (DE) of rare earth metal (REM) atom from the lowest energy structure of REMSin (n = 3–10) and their anions was calculated. The DEs of PmSin, SmSin and EuSin are nearly identical. The DEs of PmSin−, SmSin− and EuSin− are also nearly equal, and they are smaller than those of HoSin− and PrSin−.
Co-reporter:Xiaohong Xie, Dongsheng Hao, Jucai Yang
Chemical Physics 2015 Volume 461() pp:11-19
Publication Date(Web):5 November 2015
DOI:10.1016/j.chemphys.2015.08.024
•The ground-state structure of YbSin and its anion is substitutional structure.•The four DFT AEAs are in excellent agreement with the experimental data.•Theoretical AEA of 2.33 eV of YbSi9 is more reasonable than the experimental 2.60 eV.•Hardness analysis reveals that doping Yb to Sin raises photochemical sensitivity.•Relative stabilities of YbSin and their anions are examined.The structures, electron affinities, dissociation energies, hardness, and dipole moments of YbSin (n = 4–10) and their anions were examined using B3LYP, TPSSh, PBE and wB97X methods. The lowest-energy structures can be regarded as replacing a Si of the ground-state structure of Sin+1 with a Yb atom. The theoretical adiabatic electron affinities (AEAs) of YbSin are in excellent agreement with experimental data. The average absolute errors from experiment are by 0.08, 0.07, 0.05 and 0.08 eV at the B3LYP, the TPSSh, the PBE and the wB97X levels, respectively. Theoretical AEAs of 2.33 ± 0.05 eV for YbSi9 are more reliable than the experimental value of 2.60 ± 0.05 eV. The hardness analysis reveals that doping Yb atom to Sin (n = 4–10) clusters raises the photochemical sensitivity. The dissociation energies of Yb atom from YbSin and their anions were calculated to examine relative stabilities.
Co-reporter:Xiaohong Xie, Dongsheng Hao, Yuming Liu, Jucai Yang
Computational and Theoretical Chemistry 2015 Volume 1074() pp:1-8
Publication Date(Web):15 December 2015
DOI:10.1016/j.comptc.2015.10.003
•The ground-state structure of SmSin and its anion is substitutional structure.•The four DFT AEAs are in excellent agreement with the experimental data.•Hardness analysis reveals that doping Sm to Sin raises photochemical sensitivity.•Relative stabilities of SmSin and their anions are examined.•Total magnetic moments are contributed by Sm atom.The structures, adiabatic electron affinities (AEAs), dissociation energies (DEs), hardness, and magnetic moments of SmSin (n = 3–10) and their anions were examined by means of B2PLYP, TPSSh, PBE0 and B3LYP methods. Basis sets adopted are of segmented (SEG) Gaussian valence basis sets and relativistic small-core effective potentials (ECP) with additional diffuse 2dfg functions, denoted aug-SEG/ECP for Sm atoms and aug-cc-pVTZ for Si atoms. The most stable structures can be viewed as replacing a Si of the ground-state structure of Sin+1 with a Sm atom. The theoretical AEAs are in excellent agreement with experimental data, especially the AEAs at B2PLYP and TPSSh levels. The average absolute errors from experiment are by 0.04, 0.06, 0.10 and 0.11 eV at the B2PLYP, the TPSSh, the PBE0 and the B3LYP levels, respectively. The DEs of Sm from SmSin (n = 3–10) and their anions were estimated to examine their relative stabilities. Natural population analysis (NPA) reveals that the Sm atom in all the neutral and anionic clusters acts as an electron donor. Hardness analysis reveals that doping Sm atom to Sin (n = 3–10) clusters raises the photochemical sensitivity. Analysis of magnetic moments reveals that the total magnetic moments are contributed by Samarium atom.
Co-reporter:Bin Liu
Journal of Molecular Modeling 2015 Volume 21( Issue 12) pp:
Publication Date(Web):2015 December
DOI:10.1007/s00894-015-2851-6
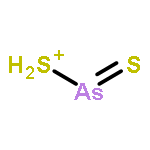
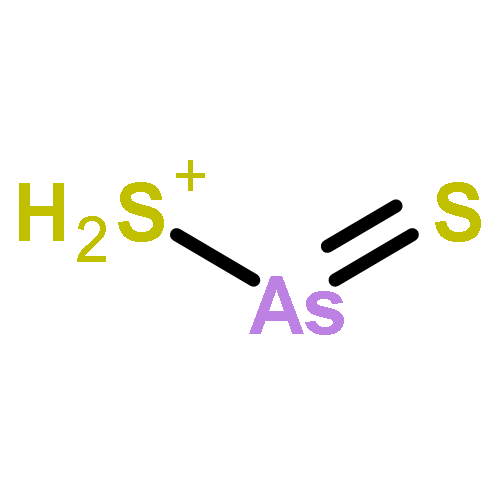
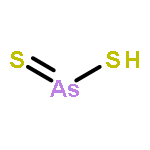
The structures and energies of neutral and charged arsenic sulfides AsnS3(−1,0,+1) (n = 1–6) were studied systematically with the G3 method. The ground-state structures of these species are reported. The ground-state structures of AsnS3 with n ≥ 4 can be considered as resulting from the replacement of an As atom of the ground-state structure of neutral Asn+1S2 by an S atom. In neutral AsnS3, the character of sulfur bonding is edge-bridging. The ground-state structures of anion AsnS3− sometimes differ from their corresponding neutral structures. In such case, they exhibit a terminal sulfur atom. The ground-state structures of cationic AsnS3+ are also sometimes different from the corresponding neutral ones. There, sulfur bonding can exhibit face-capping and arsenic can be four-fold coordinated. The potential energy surfaces of As4S3+ and As5S3+ are very flat and co-existence of various isomers of As4S3+ and As5S3+ is possible. Reliable adiabatic electron affinities (AEAs) and adiabatic ionization potentials (AIPs) of AsnS3 are predicted. There are odd–even alternations in both AEAs and AIPs as a function of size. In addition, the reliable vertical detachment energies (VDEs) and vertical ionization potentials (VIPs) are presented. The dissociation energies (DEs) of S (and/or its ion S(−/+)) from AsnS3 species and their ions were calculated to examine relative stabilities. The hardnesses and HOMO–LUMO gaps of AsnS3 (n = 1–6) were evaluated and used to discuss relative chemical reactivity.
Co-reporter:Xue Bai, Qiancheng Zhang, Aifang Gao, Jucai Yang
Computational and Theoretical Chemistry 2013 Volume 1009() pp:94-102
Publication Date(Web):1 April 2013
DOI:10.1016/j.comptc.2013.01.007
The structures, electron affinities, and dissociation energies of Asn/Asn- (n = 6–16) clusters have been examined by means of the B3LYP method. The basis set used in this examination is that of double-ζ plus polarization quality with additional diffuse s-, p-, and d-functions, designated as DZP++. Two different types of energy separation reported in this work are the adiabatic electron affinities (AEAs) and vertical detachment energies (VDEs). The ground-state structures of neutral Asn (n = 9–16) and their anions, with the exception of neutral As10, can be regarded as being derived from the ground-state structure of As7 (or As8) by attaching As(n−7) (or As(n−8)) species. And the odd-numbered Asn- clusters tend to be derived from the ground state structure of As7 by adding As(n−7) species. The reliable AEAs (VDEs) of Asn have been calculated to be 2.07 (2.18) eV for As6, 2.68 (2.77) eV for As7, 2.11 (3.09) eV for As8, 2.80 (3.08) eV for As9, 2.25 (2.51) eV for As10, 2.80 (3.23) eV for As11, 1.86 (2.11) eV for As12, 2.92 (3.02) eV for As13, 1.84 (2.16) eV for As14, 3.02 (3.10) eV for As15, and 2.13 (3.00) eV for As16. There are odd–even alternations for AEAs as a function of size of Asn, but not for VDEs. The dissociation energies (DEs) of arsenic atom from neutral Asn and their anions have been predicted to examine their relative stabilities. The results revealed that the even-numbered neutral Asn species are more stable than the odd-numbered clusters, and that the even-numbered anionic Asn- (n ⩾ 4) species are less stable than the odd-numbered species. In addition, the reliable bonding energies per atom (BEPA) of Asn have been calculated.Graphical abstractHighlights► As7 or As8 is basic unit of ground state structure of Asn/Asn- except for As10. ► There is odd-even alternation in AEA, but not in VDE. ► Theoretical AEAs and VDEs agree well with those of limited experimental values. ► DE characterized even number Asn- (n⩾4) as less stable than odd number cluster. ► Reliable bonding energy per atom of Asn has been predicted.
Co-reporter:Xue Bai, Qiancheng Zhang, Jucai Yang, and Hongmei Ning
The Journal of Physical Chemistry A 2012 Volume 116(Issue 37) pp:9382-9390
Publication Date(Web):August 30, 2012
DOI:10.1021/jp3056569
The structures and energies of neutral and charged monomethylated arsenic species CH3Asn(−1,0,+1) (n = 1–7) have been systematically investigated with the Gaussian-3 (G3) method. The ground-state structures of monomethylated arsenic species including the neutrals and the ions are vertex-methylated type. The lowest-energy structures of neutral methylated arsenic species and their ions can be viewed as being derived from corresponding to neutral and ionic arsenic clusters, respectively. The reliable electron affinities and ionization potentials of CH3Asn have been evaluated. And there are odd–even alternations in both electron affinities and ionization potentials as a function of size of CH3Asn. The dissociation energies of CH3 from neutral CH3Asn and their ions have been calculated to examine relative stabilities. The results characterized the odd-numbered neutral CH3Asn as more stable than the even-numbered systems, and the even-numbered cationic CH3Asn+ as more stable than the odd-numbered species with the exception of n = 1. The dissociation energy of CH3As+ is the maximum among all of these values. There are no odd–even alternations for anionic CH3Asn– with n ≤ 7.
Co-reporter:Hongmei Ning, Hongwei Fan, Jucai Yang
Computational and Theoretical Chemistry 2011 Volume 976(1–3) pp:141-147
Publication Date(Web):1 December 2011
DOI:10.1016/j.comptc.2011.08.015
The structures and energies of small CaSin (n = 2–10) species and their anions have been systematically investigated by means of the Gaussian-3 (G3) theory. The electron affinities have been presented and compared with those of MgSin and BeSin. The ground state structures for all of these species are found to be “substitutional structure”, which are derived from Sin+1 by replacing a Si atom with a Ca atom. The reliable adiabatic electron affinities of CaSin have been predicted to be 1.56 eV for CaSi2, 1.77 eV for CaSi3, 2.01 eV for CaSi4, 2.06 eV for CaSi5, 1.97 eV for CaSi6, 1.98 eV for CaSi7, 1.86 eV for CaSi8, 2.49 eV for CaSi9, and 2.32 eV for CaSi10. The dissociation energies of Ca atom from the lowest energy structure of CaSin clusters have been calculated in order to examine relative stabilities. Compared with those of BeSin and MgSin, the dissociation energies of Mg from MgSin are the smallest. The charge transfer of CaSin and BeSin has also been computed to further understand the interaction between the metal atom and the silicon clusters.Graphical abstract.Highlights► The ground state structure of CaSin (n = 2–10) is substitutional pattern. ► The ground state structure of their anions is also substitutional type. ► The dissociation energy of Mg from MgSin is the smallest compared to Be and Ca. ► The reliable adiabatic electron affinities of CaSin have been presented.
Co-reporter:Gang Liang, Qiang Wu, and Jucai Yang
The Journal of Physical Chemistry A 2011 Volume 115(Issue 29) pp:8302-8309
Publication Date(Web):June 23, 2011
DOI:10.1021/jp203585p
The structures and energies of Asn (n = 2–8) neutrals, anions, and cations have been systematically investigated by means of the G3 schemes. The electron affinities, ionization potentials, binding energies, and several dissociation energies have been calculated and compared with limited experimental values. The results revealed that the potential surfaces of neutral Asn clusters are very shallow, and two types of structural patterns compete with each other for the ground-state structure of Asn with n ≥ 6. One type is derived from the benzvalene form of As6, and another is derived from the trigonal prism of As6. The previous photoelectron spectrum (taken from J. Chem. Phys. 1998, 109, 10727) for As3 has been reassigned in light of the G3 results. The experimental electron affinities of As3 were measured to be 1.81 eV, not 1.45 eV. We inferred from the conclusion of G3 and density functional theory that the experimental electron affinities of 1.7 and 3.51 eV for As5 are unreliable. The reliable electron affinities were predicted to be 0.83 eV for As2, 1.80 eV for As3, 0.54 eV for As4, 3.01 eV for As5, 2.08 eV for As6, 2.93 eV for As7, and 2.02 eV for As8. The G3 ionization potentials were calculated to be 9.87 eV for As2, 7.33 eV for As3, 8.65 eV for As4, 6.68 eV for As5, 7.97 eV for As6, 6.58 eV for As7, and 7.65 eV for As8. The binding energies per atom were evaluated to be 1.99 eV for As2, 2.01 eV for As3, 2.61 eV for As4, 2.39 eV for As5, 2.51 eV for As6, 2.55 eV for As7, and 2.67 eV for As8. These theoretical values of As2, As3, and As4 are in excellent agreement with those of experimental results. Several dissociation energies were carried out to examine relative stabilities. This characterized the even-numbered clusters as more stable than the odd-numbered species.
Co-reporter:Hongwei Fan, Zhiqing Ren, Jucai Yang, Dongsheng Hao, Qiancheng Zhang
Journal of Molecular Structure: THEOCHEM 2010 Volume 958(1–3) pp:26-32
Publication Date(Web):30 October 2010
DOI:10.1016/j.theochem.2010.07.022
The equilibrium geometries, energies, charge transfer, and dipole moments of small MgSin (n = 2–10) species and their anions have been systematically investigated at the highest level of Gaussian-3 (G3) theory. For neutral MgSin clusters, the ground-state structures are found to be “attaching structure” in which the Mg atom is bound to Sin clusters. The lowest-energy structures for their anions, however, are found to be “substitutional structures”, which are derived from Sin+1 by replacing a Si atom with a Mg atom. The reliable adiabatic electron affinities of MgSin have been predicted to be 1.84 eV for MgSi2, 1.90 eV for MgSi3, 2.17 eV for MgSi4, 2.35 eV for MgSi5, 2.45 eV for MgSi6, 2.18 eV for MgSi7, 2.98 eV for MgSi8, 3.00 eV for MgSi9, and 2.00 eV for MgSi10. The dissociation energies of Mg atom from the lowest-energy structure of MgSin clusters have been evaluated to examine relative stabilities. The charge transfer and dipole moments have also been calculated to further understand the interaction between the Mg atom and the silicon clusters.
Co-reporter:Caixia Dong, Jucai Yang, Hongmei Ning, Chunping Li
Journal of Molecular Structure: THEOCHEM 2010 Volume 950(1–3) pp:64-71
Publication Date(Web):30 June 2010
DOI:10.1016/j.theochem.2010.03.025
The structures of the As–nucleobases (As–adenine, –guanine, –cytosine, –thymine, and –uracil) and their anions are studied at the density functional B3LYP with a double-ζ plus diffuse (DZP++) basis set. The lowest energy structures of these complexes are presented. For the case of neutral systems, a strong association between arsenic atom and nucleobases is found and the ground state structures are R–As–H (R = nucleobases radical) complexes. For anions, the most stable structures are also R–As–H forms, but their association sites are different from corresponding neutrals with the exception of As–adenine. The extra electron of the anions is mainly localized on the arsenic atom. The adiabatic electron affinity (AEA), the vertical electron affinity (VEA), and the vertical detachment energy (VDE) for these complexes are reported. The formation energies of these complexes are also estimated, respectively, in order to examine relative stabilities. The stability order is As–guanine > As–adenine > As–uracil > As–cytosine > As–thymine.
Co-reporter:Yousuo Zhang, Jucai Yang, Hongwei Fan, Chunping Li
Journal of Molecular Structure: THEOCHEM 2010 Volume 951(1–3) pp:21-27
Publication Date(Web):15 July 2010
DOI:10.1016/j.theochem.2010.03.038
The hydrogenated pyridoxine (PN) species in cationic, neutral, and anionic states have been studied. The structures, energetics, and theoretical electron affinities are predicted by means of the B3LYP/DZP++ method. These radical and anionic species come from consecutive electron attachment to the corresponding protonated (PN+H)+ cations in low pH environments. The most reliable proton affinities of PN (protonation at N1) are predicted to be 232.3 kcal/mol. The radicals range within 11.5 kcal/mol in relative energy and radicals r1 (attached H to N1 site of PN) are the lowest energy. Structures r7 and r8 are predicted to extrude water molecule. The theoretical adiabatic electron affinities (AEA), vertical electron affinities (VEA), and vertical detachment energies (VDE) are estimated. The ranges of AEA for six radicals are from 0.44 (or 0.35) to 1.76 eV. Taking into account the solvent effect, protonation of PN in aqueous solution is more stable and neutral hydrogenated PN more readily takes up solvated electrons.
Co-reporter:Hongyu Li, Chunping Li, Hongwei Fan, Jucai Yang
Journal of Molecular Structure: THEOCHEM 2010 Volume 952(1–3) pp:67-73
Publication Date(Web):30 July 2010
DOI:10.1016/j.theochem.2010.04.023
A theoretical study of the equilibrium geometries, spin multiplicities, dipole moments, electron affinities, and mean bond dissociation energies of neutral Mn(Bz)m (M = Ti, V, Cr, n ≤ 2, m ≤ 2, Bz = benzene) species and their anions is carried out by means of B3LYP density functional method. The ground state geometries are presented. The spin multiplicities of MBz, M2Bz, M(Bz)2 and M2(Bz)2 systems are very different from each other. The adiabatic electron affinity (AEA) with and without ZPVE correction, the vertical electron affinity (VEA), and the vertical detachment energy (VDE) of these complexes are evaluated. The theoretical AEA is in agreement with the limited experimental data. The mean bond dissociation energies for all of these neutral complexes are also calculated to examine their relative stabilities. The order of stability of Mn(Bz)m (M = Ti, V, Cr, n ≤ 2, m ≤ 2, Bz = benzene) species is M(Bz)2 > M2(Bz)2 > M2Bz > MBz and Tin(Bz)m > Vn(Bz)m > Crn(Bz)m.
Co-reporter:Hongwei Fan, Jucai Yang, Wei Lu, Hongmei Ning and Qiancheng Zhang
The Journal of Physical Chemistry A 2010 Volume 114(Issue 2) pp:1218-1223
Publication Date(Web):December 18, 2009
DOI:10.1021/jp910326a
The equilibrium geometries and energies of neutral BeSin (n = 2-10) species and their anions have been studied at the highest level of Gaussian-3 (G3) theory. The results reveal that the ground-state structures of these clusters are Be-encapsulated in silicon cages with n ≥ 8. The reliable adiabatic electron affinities of BeSin have been predicted to be 1.68 eV for BeSi2, 1.87 eV for BeSi3, 2.33 eV for BeSi4, 2.29 eV for BeSi5, 2.11 eV for BeSi6, 2.37 eV for BeSi7, 2.95 eV for BeSi8, 2.74 eV for BeSi9, and 1.92 eV for BeSi10. The dissociation energies of Be atom from BeSin, Si atom from BeSin, and Si atom from Sin clusters have also been calculated, respectively, to examine relative stabilities. The trend of stability of BeSin changed with n is converse to that of Sin when n ≤ 7. From n ≥ 8, the encapsulated Be atom in silicon cages not only results in an identical trend for stability of BeSin and Sin but also improves the stability of Sin clusters.
Co-reporter:Dong-Sheng Hao;Jin-Rong Liu;Wen-Guang Wu;Ju-Cai Yang
Theoretical Chemistry Accounts 2009 Volume 124( Issue 5-6) pp:
Publication Date(Web):2009 November
DOI:10.1007/s00214-009-0635-8
The neutral SinK (n = 2–8) clusters and their anions have been systematically studied by means of the higher level of Gaussian-3 schemes. Equilibrium geometries and electron affinities have been calculated and are discussed for each considered size. For neutral SinK clusters, the ground state structure is found to be “attaching structure”, in which the K atom is bound to Sin clusters. The most stable isomer for their anions, however, is found to be “substitutional structures”, which is derived from Si(n+1) by replacing the Si atom with a K. The dissociation energies of K atom from the lowest energy structures of SinK have also been estimated to examine relative stabilities.
Co-reporter:Dongsheng Hao, Jinrong Liu and Jucai Yang
The Journal of Physical Chemistry A 2008 Volume 112(Issue 41) pp:10113-10119
Publication Date(Web):September 23, 2008
DOI:10.1021/jp804393k
The molecular structures of neutral SinLi (n = 2−8) species and their anions have been studied by means of the higher level of the Gaussian-3 (G3) techniques. The lowest energy structures of these clusters have been reported. The ground-state structures of neutral clusters are “attaching structures”, in which the Li atom is bound to Sin clusters. The ground-state geometries of anions, however, are “substitutional structures”, which is derived from Sin+1 by replacing a Si atom with a Li−. The electron affinities of SinLi and Sin have been presented. The theoretical electron affinities of Sin are in good agreement with the experiment data. The reliable electron affinities of SinLi are predicted to be 1.87 eV for Si2Li, 2.06 eV for Si3Li, 2.01 eV for Si4Li, 2.61 eV for Si5Li, 2.36 eV for Si6Li, 2.21 eV for Si7Li, and 3.18 eV for Si8Li. The dissociation energies of Li atom from the lowest energy structures of SinLi and Si atom from Sin clusters have also been estimated respectively to examine relative stabilities.
Co-reporter:LiHua Lin, JuCai Yang, HongMei Ning, DongSheng Hao, HongWei Fan
Journal of Molecular Structure: THEOCHEM 2008 Volume 851(1–3) pp:197-206
Publication Date(Web):28 February 2008
DOI:10.1016/j.theochem.2007.11.014
The molecular structures, electron affinities, and dissociation energies of the SinNa/SinNa− (n ⩽ 10) species have been investigated by means of five hybrid and pure density functional theory (DFT) methods. The basis sets used in this work is of double-ζ plus polarization quality with additional diffuse functions, denoted DZP++. The geometries are fully optimized with each DFT method independently. Three different types of the neutral-anion energy separations presented in this work are the adiabatic electron affinity (EAad), the vertical electron affinity (EAvert), and the vertical detachment energy (VDE). Compared with the limited experimental values of EAad and VDE, the BPW91 and B3PW91 scheme provided the most reliable EAad and VDE values. The dissociation energy De (SinNa → Sin+Na) for the neutral SinNa and De(SinNa-→Sin-+Na) for the anionic SinNa− species have also been reported and used to understand relative stability.
Co-reporter:Ju-Cai Yang;Lihua Lin;Yousuo Zhang;Abraham F. Jalbout
Theoretical Chemistry Accounts 2008 Volume 121( Issue 1-2) pp:83-90
Publication Date(Web):2008 September
DOI:10.1007/s00214-008-0452-5
The molecular structures, electron affinities, and dissociation energies of neutral SinLi (n = 2–10) species and their anions have been studied by the B3LYP and the BPW91 methods in conjunction with a DZP++ basis set. The geometries have been fully optimized with each of the proposed methods. The ground state structure of neutral SinLi keeps the corresponding Sin framework unchanged. For anion, the corresponding Sin (or \({{\rm Si}_{n}^{-}}\)) framework changes largely when n ≥ 7. To evaluate the stability of the resulting anions we have calculated the adiabatic electron affinity (EAad), the vertical electron affinity (EAvert), and the vertical detachment energy (VDE). The dissociating energies of Li from the lowest energy structures of neutral SinLi and their anions are calculated to examine relative stabilities.
Co-reporter:Xue Bai, HongMei Ning, JuCai Yang, HongWei Fan, DongSheng Hao, CaiLing Li
Journal of Molecular Structure: THEOCHEM 2007 Volume 808(1–3) pp:41-52
Publication Date(Web):30 April 2007
DOI:10.1016/j.theochem.2006.12.037
The geometries and energies of SinH2 (n = 3, 5–10) have been systematically investigated by means of MP2/6-311++G∗∗//MP2/6-31G∗∗ and B3LYP/6-311++G∗∗ schemes. Several geometric arrangements have been considered for each cluster. All the geometries considered have been completely optimized within the given symmetry constrains. The results show that the ground state geometries of Si4H2, Si6H2, and Si8H2 are attaching two H-atoms to one Si-atom and others are bonding two H-atoms to two Si-atoms. The results of the lowest energy structure of SinH2 at MP2 levels are the same as those of results at B3LYP levels with the exception of Si5H2. However, the MP4(SDQ) result is the same as the B3LYP for Si5H2. At B3LYP level of theory, dissociation energies of the lowest energy structures of SinH2 (n = 3–10) have been computed and used to understand relative stability. Other properties, such as HOMO-LUMO gap, hardness, vertical electron affinities, and vertical ionization potential have been assessed.
Co-reporter:HongMei Ning, JuCai Yang, LiHua Lin, DongSheng Hao, HongWei Fan
Journal of Molecular Structure: THEOCHEM 2007 Volume 816(1–3) pp:137-144
Publication Date(Web):20 August 2007
DOI:10.1016/j.theochem.2007.04.009
The molecular structures and electron affinities of the SinH2/SinH2- (n = 5–10) species have been examined using three density functional theory (DFT) methods. The basis set used in this work is of double-ζ plus polarization quality with additional diffuse s- and p-type functions, denoted DZP++. The geometries are fully optimized with each DFT method independently. Four different types of the neutral-anion energy separations presented in this work are the adiabatic electron affinity (EAad), zero-point vibrational energies corrected EAad (EAzero), the vertical electron affinity (EAvert), and the vertical detachment energy (VDE). The first Si–H dissociation energies De (SinH2 → Sin + H) for neutral SinH2 and De (SinH2-→SinH-+H) for anionic SinH2- species have also been presented.
Co-reporter:JuCai Yang, WenGuo Xu, WenSheng Xiao
Journal of Molecular Structure: THEOCHEM 2005 Volume 719(1–3) pp:89-102
Publication Date(Web):14 April 2005
DOI:10.1016/j.theochem.2004.12.035
The silicon clusters structures, electron affinities, and dissociation energies of the Sin/Sin− (n=2–10) species have been examined using seven hybrid and pure density functional theory (DFT) methods. The basis set used in this work is of double-ζ plus polarization quality with additional diffuse s- and p-type functions, denoted DZP++. The geometries are fully optimized with each DFT method independently. Four different types of energy separations presented in this work are the adiabatic electron affinity (EAad), zero-point vibrational energies (ZPVE) corrected EAad (EAzero), the vertical electron affinity (EAvert), and the vertical detachment energy (VDE). The first Si–Si dissociation energies De (Sin→Sin−1+Si) for Sin, and both De(Sin−→Sin−1+Si−) and (Sin−→Sin−1−+Si) for Sin− species have also been reported. The most reliable adiabatic electron affinities, obtained at the DZP++ BPW91 level of theory, are 2.16 (2.15) eV for Si2, 2.32 (2.32) eV for Si3, 2.24 (2.25) eV for Si4, 2.51 (2.51) eV for Si5, 2.11 (2.12) eV for Si6, 2.06 (2.07) eV for Si7, 2.86 (2.85) eV for Si8, 2.28 (2.28) eV for Si9, 2.45 (2.46) eV for Si10. (EAzero values are in parentheses). While BP86, B3P86 and BPW91 predict to the most reliable dissociation energies. The dissociation energies for Sin→Sin−1+Si are predicted to be 3.26 (3.23) eV for Si2, 3.96 (3.92) eV for Si3, 4.39 (4.33) eV for Si4, 3.68 (3.62) eV for Si5, 4.12 (4.08) eV for Si6, 4.07 (4.01) eV for Si7, 2.76 (2.73) eV for Si8, 4.28 (4.22) eV for Si9, 4.33 (4.28) eV for Si10 with error of 0.13 (0.16) eV (corrected with ZPVE in parentheses). And the dissociation energies of Sin−→Sin−1−+Si are predicted to be 3.95 (3.92) eV for Si2−, 4.14 (4.11) eV for Si3−, 4.29 (4.24) eV for Si4−, 3.98 (3.92) eV for Si5−, 3.72 (3.68) eV for Si6−, 4.01 (3.96) eV for Si7−, 3.59 (3.54) eV for Si8−, 3.69 (3.63) eV for Si9−, and 4.51 (4.46) eV for Si10−.