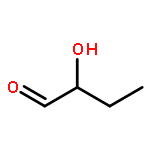
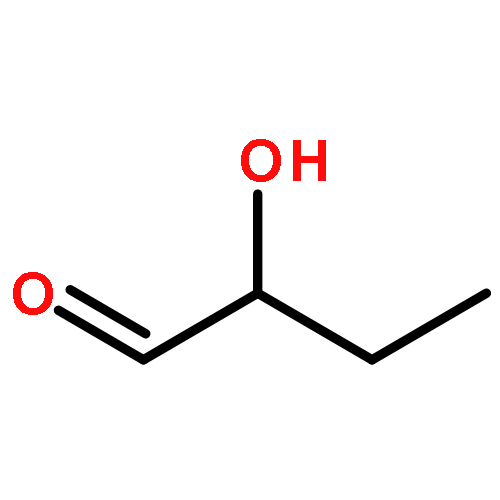
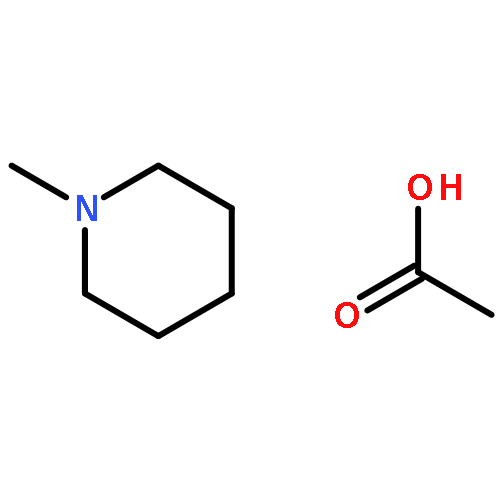
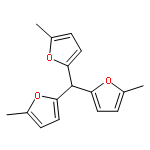
Co-reporter:N. Hansen;R. Krishna;J. M. van Baten;A. T. Bell;F. J. Keil
The Journal of Physical Chemistry C January 8, 2009 Volume 113(Issue 1) pp:235-246
Publication Date(Web):2017-2-22
DOI:10.1021/jp8073046
A continuum model based on the Maxwell−Stefan (M-S) equations in combination with the ideal adsorbed solution theory has been used to analyze the influence of adsorption thermodynamics and intraparticle diffusional transport on the overall kinetics of benzene alkylation with ethene over H-ZSM-5. The parameters appearing in the M-S equations were obtained from molecular dynamics simulations, and pure component adsorption isotherms were obtained from configurational-bias Monte Carlo simulations in the grand canonical ensemble. Rate coefficients for the elementary steps of the alkylation were taken from quantum chemical calculations. The intrinsic kinetics of two different reaction schemes were analyzed. The simulations show that all apparent rate parameters of the alkylation are strongly dependent on the reaction conditions. By taking diffusional limitation into account, experimentally determined reaction rates and the orders in the partial pressures of reactants can be reproduced. The results of this study show that empirical power law rate expressions become inappropriate when used to correlate kinetic data over a broad range of conditions. In addition, it is demonstrated that the usual approaches to determine effectiveness factors for reactions in porous media, which assume a constant effective diffusivity, may lead to substantial deviations from rigorous simulations, whereas the simulation model developed here can be used to predict the effectiveness factor for zeolite particles for any set of reaction conditions.
Co-reporter:Amber JandaBess Vlaisavljevich, Berend SmitLi-Chiang Lin, Alexis T. Bell
The Journal of Physical Chemistry C 2017 Volume 121(Issue 3) pp:
Publication Date(Web):December 20, 2016
DOI:10.1021/acs.jpcc.6b09703
Monte Carlo simulations are used to systematically investigate the effects of structural topology on the thermodynamics of n-alkanes adsorbed at Brønsted protons in zeolites having one-dimensional channel systems. In zeolites without cages, the enthalpy and entropy of adsorption (ΔHads-H+ and ΔSads-H+) at fixed pore-limiting diameter (PLD) generally increase (become less negative) as the ratio of the minimum to maximum channel diameter decreases, and are lowest when this ratio equals 1 (corresponding to approximately circular cross sections). The effect of a change in diameter ratio on the free energy of adsorption (ΔAads-H+) is weak because the changes in ΔHads-H+ and TΔSads-H+ largely cancel. The addition of cages having a largest-cavity diameter (LCD) greater than the PLD increases both ΔHads-H+ and ΔSads-H+. Replacing channels with cages of the same diameter does not change ΔSads-H+ significantly when the PLD is similar to the alkane length but decreases both ΔHads-H+ and ΔAads-H+ because of the greater surface area of cages relative to channels. The selectivity to adsorption via a central C–C bond vs a terminal bond when cages are absent is smallest for PLDs near the alkane length and, when cages are present, is even lower when the LCD exceeds the alkane length. This effect is attributed to more rotation of the alkane in cages vs channels. The results show that ΔAads-H+ at 773 K can be tuned by manipulating a characteristic dimension (LCD, PLD) and topology (e.g., adding cages) simultaneously, in order to circumvent the compensating changes in TΔSads-H+ and ΔHads-H+ that occur upon changing only one structural parameter.
Co-reporter:Amber Janda; Bess Vlaisavljevich; Li-Chiang Lin; Berend Smit
Journal of the American Chemical Society 2016 Volume 138(Issue 14) pp:4739-4756
Publication Date(Web):February 24, 2016
DOI:10.1021/jacs.5b11355
The effects of zeolite structure on the kinetics of n-butane monomolecular cracking and dehydrogenation are investigated for eight zeolites differing in the topology of channels and cages. Monte Carlo simulations are used to calculate enthalpy and entropy changes for adsorption (ΔHads-H+ and ΔSads-H+) of gas-phase alkanes onto Brønsted protons. These parameters are used to extract intrinsic activation enthalpies (ΔHint‡), entropies (ΔSint‡), and rate coefficients (kint) from measured data. As ΔSads-H+ decreases (i.e., as confinement increases), ΔHint‡ and ΔSint‡ for terminal cracking and dehydrogenation decrease for a given channel topology. These results, together with positive values observed for ΔSint‡, indicate that the transition states for these reactions resemble products. For central cracking (an earlier transition state), ΔHint‡ is relatively constant, while ΔSint‡ increases as ΔSads-H+ decreases because less entropy is lost upon protonation of the alkane. Concurrently, selectivities to terminal cracking and dehydrogenation decrease relative to central cracking because ΔSint‡ decreases for the former reactions. Depending on channel topology, changes in the measured rate coefficients (kapp) with confinement are driven by changes in kint or by changes in the adsorption equilibrium constant (Kads-H+). Values of ΔSint‡ and ΔHint‡ are positively correlated, consistent with weaker interactions between the zeolite and transition state and with the greater freedom of movement of product fragments within more spacious pores. These results differ from earlier reports that ΔHint‡ and ΔSint‡ are structure-insensitive and that kapp is dominated by Kads-H+. They also suggest that ΔSads-H+ is a meaningful descriptor of confinement for zeolites having similar channel topologies.
Co-reporter:Gregory R. Johnson and Alexis T. Bell
ACS Catalysis 2016 Volume 6(Issue 1) pp:100
Publication Date(Web):November 17, 2015
DOI:10.1021/acscatal.5b02205
The effects of Zr promotion on the structure and performance of Co-based Fischer–Tropsch synthesis (FTS) catalysts were investigated. Inclusion of Zr in the catalysts was found to increase the FTS turnover frequency and the selectivity to C5+ hydrocarbons and to decrease the selectivity to methane under most operating conditions. These improvements to the catalytic performance are a function of Zr loading up to an atomic ratio of Zr/Co = 1.0, above which the product selectivity is insensitive to higher concentrations of the promoter. Characterization of the Co nanoparticles by different methods demonstrated that the optimal Zr loading corresponds to half monolayer coverage of the Co surface by the promoter. Measurements of the rate of FTS at different pressures and temperatures established that the kinetics data for both the Zr-promoted and unpromoted catalysts are described by a two-parameter Langmuir–Hinshelwood expression. The parameters used to fit this rate law to the experimental data indicate that the apparent rate coefficient and the CO adsorption constant for the Zr-promoted catalysts are higher than those for the unpromoted catalyst. Elemental mapping by means of STEM-EDS provided evidence that Zr is highly dispersed over the catalyst surface and has limited preference for association with the Co nanoparticles. In situ X-ray absorption spectroscopy confirmed the absence of mixing between the Zr and Co in the nanoparticles. These results suggest that Zr exists as a partial layer of ZrO2 on the surface of the Co metal nanoparticles. Accordingly, it is proposed that Zr promotion effects originate from sites of enhanced activity at the interface between Co and ZrO2. The possibility that ZrO2 acts as a Lewis acid to assist in CO dissociation as well as to increase the ratio of CO to H adsorbed on the catalyst surface is discussed.Keywords: cobalt; Fischer−Tropsch synthesis; heterogeneous catalysis; promotion; zirconium
Co-reporter:John G. Howell, Yi-Pei Li, and Alexis T. Bell
ACS Catalysis 2016 Volume 6(Issue 11) pp:7728
Publication Date(Web):October 6, 2016
DOI:10.1021/acscatal.6b01842
A detailed investigation was conducted on the factors influencing the properties of silica-supported tungsten oxide catalysts for propene metathesis. A principal goal of this work was to identify the processes involved in the formation of catalytically active sites. To probe the influence of dispersion, samples were prepared across a range of W loadings using two methods of catalyst preparation: incipient wetness impregnation of amorphous silica and ion exchange of mesoporous SBA-15. The samples were characterized by nitrogen adsorption, UV–vis, Raman, and X-ray absorption spectroscopy (XAS). Catalytic activity was observed to increase with W surface concentration up to the point where WO3 nanoparticles formed. The catalytic performance of all samples was enhanced 2-fold by pretreatment in He, in comparison to pretreatment in air. In situ characterization of samples pretreated in He by Raman and XAS shows an increase in the relative concentration of isolated dioxo W(6+) species relative to mono-oxo W(6+) species, and in situ XAS data collected during propene metathesis indicated that a similar conversion occurs for air-pretreated samples in the presence of propene. For both air- and He-pretreated catalysts an activation period was observed, during which the activity increased and attained steady-state activity. This period was significantly longer for air-pretreated catalysts and was accompanied by the transient formation of acetone. While acetone was not observed during the much shorter transient of He-pretreated samples, in situ XAS provided evidence of reduction occurring in these samples upon contact with propene. It is also notable that, independent of the manner of catalyst preparation or pretreatment, the rate of propene metathesis is first order in propene and exhibits an activation energy of 200 kJ/mol. A model is proposed to explain why only a fraction of the isolated tungstate species is active for propene metathesis (∼5%) and why this fraction increases with increasing concentration of W dispersed on silica.Keywords: butene; catalyst activation; metathesis; propene; silica; tungstate species
Co-reporter:Christopher R. Ho, Sankaranarayanapillai Shylesh, and Alexis T. Bell
ACS Catalysis 2016 Volume 6(Issue 2) pp:939
Publication Date(Web):December 23, 2015
DOI:10.1021/acscatal.5b02672
The mechanism and kinetics for ethanol coupling to n-butanol over hydroxyapatite (HAP) were investigated at 573–613 K. In situ titration experiments show that the active sites for acetaldehyde and butanol formation are different. In combination with FTIR studies, it was found that ethanol dehydrogenation is catalyzed by Ca–O sites, whereas condensation of acetaldehyde is catalyzed by CaO/PO43– pairs. Measurements of the reaction kinetics at various ethanol (3.5–9.4 kPa) and acetaldehyde (0.055–0.12 kPa) partial pressures reveal that direct condensation involving two ethanol molecules does not play a significant role in butanol formation; instead, n-butanol is formed via a Guerbet pathway. At a constant acetaldehyde pressure, enolate formation is rate-limiting, and ethanol inhibits acetaldehyde condensation rates by competitive adsorption. A model of the reaction kinetics consistent with all experimental observations is developed.Keywords: acetaldehyde; butanol; ethanol coupling; Guerbet reaction; hydroxyapatite
Co-reporter:Sankaranarayanapillai Shylesh, Daeyoup Kim, Amit A. Gokhale, Christian G. Canlas, Jochem O. Struppe, Christopher R. Ho, Deepak Jadhav, Alice Yeh, and Alexis T. Bell
Industrial & Engineering Chemistry Research 2016 Volume 55(Issue 40) pp:10635
Publication Date(Web):September 22, 2016
DOI:10.1021/acs.iecr.6b03601
The effects of chemical composition and pretreatment on Mg–Al hydrotalcites and alumina-supported MgO were evaluated for the gas-phase, self-condensation reaction of C3–C5 biomass-derived methyl ketones. We show that the selectivity toward the acyclic dimer enone and the cyclic enone trimer can be tuned by controlling the temperature of hydrotalcite calcination. Methyl ketone cyclization is promoted by Lewis acidic sites present on the hydrotalcite catalysts. XRD and thermal decomposition analysis reveal that the formation of periclase MgO starts above 623 K accompanied by complete disappearance of the hydrotalcite structure and is accompanied by an increase in hydroxyl condensation as the formation of well-crystallized periclase. 27Al MQMAS and 25Mg MAS NMR show that at progressively higher temperatures, Al3+ cations diffuses out of the octahedral brucite layers and incorporate into the tetrahedral and octahedral sites of the MgO matrix thereby creating defects to compensate the excess positive charge generated. The oxygen anions adjacent to the Mg2+/Al3+ defects become coordinatively unsaturated, leading to the formation of new basic sites. A kinetic isotope effect, kH/kD = 0.96, is observed at 473 K for the reaction of (CH3)2CO versus (CD3)2CO, which suggests that carbon–carbon bond formation leading to the dimer aldol product is the rate-determining step in the condensation reaction of methyl ketones. We also show that acid–base catalysts having similar reactivity and higher hydrothermal stability to that of calcined hydrotalcites can be achieved by creating defects in MgO crystallites supported alumina as a consequence of the diffusion of Al3+ cations into MgO. The physical properties of these materials are shown to be very similar to those of hydrotalcite calcined at 823 K.
Co-reporter:Dr. Shannon Klaus;Dr. Lena Trotochaud;Dr. Mu-Jeng Cheng; Martin Head-Gordon; Alexis T. Bell
ChemElectroChem 2016 Volume 3( Issue 1) pp:66-73
Publication Date(Web):
DOI:10.1002/celc.201500364
Abstract
Addition of Fe to Ni- and Co-based (oxy)hydroxides has been shown to enhance the activity of these materials for electrochemical oxygen evolution. Here we show that Fe cations bound to the surface of oxidized Au exhibit enhanced oxygen evolution reaction (OER) activity. We find that the OER activity increases with increasing surface concentration of Fe. Density functional theory analysis of the OER energetics reveals that oxygen evolution over Fe cations bound to a hydroxyl-terminated oxidized Au (Fe–Au2O3) occurs at an overpotential ∼0.3 V lower than over hydroxylated Au2O3 (0.82 V). This finding agrees well with experimental observations and is a consequence of the more optimal binding energetics of OER reaction intermediates at Fe cations bound to the surface of Au2O3. These findings suggest that the enhanced OER activity reported recently upon low-potential cycling of Au may be due to surface Fe impurities rather than to “superactive” AuIII surfaquo species.
Co-reporter:Dr. Shannon Klaus;Dr. Lena Trotochaud;Dr. Mu-Jeng Cheng; Martin Head-Gordon; Alexis T. Bell
ChemElectroChem 2016 Volume 3( Issue 1) pp:
Publication Date(Web):
DOI:10.1002/celc.201500527
Co-reporter:Gregory R. Johnson, Sebastian Werner, and Alexis T. Bell
ACS Catalysis 2015 Volume 5(Issue 10) pp:5888
Publication Date(Web):August 27, 2015
DOI:10.1021/acscatal.5b01578
Mn is an effective promoter for improving the activity and selectivity of Co-based Fischer–Tropsch synthesis (FTS) catalysts, but the mechanism by which this promoter functions is poorly understood. The work reported here was aimed at defining the manner in which Mn interacts with Co and determining how these interactions affect the activity and selectivity of Co. Detailed measurements are reported for the kinetics of FTS as a function of Mn/Co ratio, temperature, and reactant partial pressure. These data are described by a single, two-parameter rate expression. Mn promotion was found to increase both the apparent rate constant for CO consumption and the CO adsorption constant. Further evidence for enhanced CO adsorption and dissociation was obtained from measurements of temperature-programmed desorption of CO and CO disproportionation rates, respectively. Quantitative analysis of elemental maps obtained by STEM-EDS revealed that the promoter accumulates preferentially on the surface of Co nanoparticles at low Mn loadings, resulting in a rapid onset of improvements in the product selectivity as the Mn loading increases. For catalysts prepared with loadings higher than Mn/Co = 0.1, the additional Mn accumulates in the form of nanometer-scale particles of MnO on the support. In situ IR spectra of adsorbed CO show that Mn promotion increases the abundance of adsorbed CO with weakened C–O bonds. It is proposed that the cleavage of the C–O bond is promoted through Lewis acid–base interactions between the Mn2+ cations located at the edges of MnO islands covering the Co nanoparticles and the O atom of CO adsorbates adjacent to the MnO islands. The observed decrease in selectivity to CH4 and the increased selectivity to C5+ products with increasing Mn/Co ratio are attributed to a decrease in the ratio of adsorbed H to CO on the surface of the supported Co nanoparticles.Keywords: cobalt; Fischer−Tropsch synthesis; heterogeneous catalysis; manganese; promotion
Co-reporter:Eric R. Sacia, Matthew H. Deaner, Ying “Lin” Louie and Alexis T. Bell
Green Chemistry 2015 vol. 17(Issue 4) pp:2393-2397
Publication Date(Web):23 Jan 2015
DOI:10.1039/C4GC02292K
A novel approach to produce biomass-derived gasoline is the hydrolysis of 2,5-dimethylfuran (DMF) to produce 2,5-hexanedione followed by base-catalyzed intramolecular aldol condensation of this product to form 3-methylcyclopent-2-enone (MCP). By proper choice of catalysts and conditions, MCP yields of 98% can be achieved. We further show that hydrogenation of MCP over Pt/NbOPO4 gives methylcyclopentane with virtually quantitative yields. Methylcyclopentane is an attractive gasoline substitute for ethanol, since its octane number is similar to ethanol and its gravimetric energy density is 58% higher.
Co-reporter:Kristopher R. Enslow and Alexis T. Bell
Catalysis Science & Technology 2015 vol. 5(Issue 5) pp:2839-2847
Publication Date(Web):16 Mar 2015
DOI:10.1039/C5CY00077G
A number of Lewis acid catalysts were screened for their effectiveness in converting both xylose and glucose in aqueous media to furfural and 5-HMF, respectively. While other catalysts were found to be more active, SnCl4 was identified as the most selective Lewis acid. Hydrolysis of SnCl4 was observed at various concentrations and temperatures resulting in the production of Brønsted acidic protons in a 3.5:1 ratio to Sn4+ at all SnCl4 concentrations above 60 °C. As a consequence, there was no need to add a Brønsted acid in order to promote the dehydration of either xylose or glucose. SnCl4-promoted isomerization/dehydration of xylose and glucose at 140 °C in water resulted in conversions of 55% and 33%, respectively, after 2 h of reaction, and furfural and 5-HMF selectivities of up to 58% and 27%, respectively. Significant conversion of sugars to humins was observed in both cases, and in the case of glucose, degradation of 5-HMF to levulinic and formic acids was also noted. The effects of secondary reactions could be greatly suppressed by extraction of the furanic product as it was produced. Using n-butanol as the extracting agent, xylose and glucose conversions of 90% and 75%, respectively, were observed after 5 h of reaction, and the selectivities to furfural and 5-HMF increased to 85% and 69%, respectively. Small additional increases in the furfural and 5-HMF selectivities were obtained by adding LiCl to the aqueous phase without much effect on the conversion of either sugar. In this case, the selectivities to furfural and 5-HMF were 88% and 72%, respectively, after 5 h of reaction at 140 °C.
Co-reporter:Shannon Klaus
The Journal of Physical Chemistry C 2015 Volume 119(Issue 13) pp:7243-7254
Publication Date(Web):March 3, 2015
DOI:10.1021/acs.jpcc.5b00105
Ni-(oxy)hydroxide-based materials are promising earth-abundant catalysts for electrochemical water oxidation in basic media. Recent findings demonstrate that incorporation of trace Fe impurities from commonly used KOH electrolytes significantly improves oxygen evolution reaction (OER) activity over NiOOH electrocatalysts. Because nearly all previous studies detailing structural differences between α-Ni(OH)2/γ-NiOOH and β-Ni(OH)2/β-NiOOH were completed in unpurified electrolytes, it is unclear whether these structural changes are unique to the aging phase transition in the Ni-(oxy)hydroxide matrix or if they arise fully or in part from inadvertent Fe incorporation. Here, we report an investigation of the effects of Fe incorporation on structure–activity relationships in Ni-(oxy)hydroxide. Electrochemical, in situ Raman, X-ray photoelectron spectroscopy, and electrochemical quartz crystal microbalance measurements were employed to investigate Ni(OH)2 thin films aged in Fe-free and unpurified (reagent-grade) 1 M KOH (<1 ppm Fe). We find that Ni films aged in unpurified electrolyte can incorporate ≥20% Fe after 5 weeks of aging, and the maximum catalyst activity is comparable to that reported for optimized Ni1–xFexOOH catalysts. Conversely, Fe-free Ni(OH)2 films exhibit a substantially lower activity and higher Tafel slope for the OER. Films aged in Fe-free electrolyte are predominantly disordered β-Ni(OH)2/β-NiOOH if maintained below 0.7 V vs Hg/HgO in 1 M KOH and will “overcharge” to form a mixture of γ- and β-NiOOH above this potential. Fe-containing Ni(OH)2 films evidence a lesser extent of β-Ni(OH)2 formation and instead exhibit NiOOH structural changes in accordance with the formation of a NiFe-layered double hydroxide phase. Furthermore, turnover frequency calculations indicate that Fe is the active site within this phase, and above ∼11% Fe content, a separate, Fe-rich phase forms. These findings are the first to demonstrate the in situ changes in the catalyst structure resulting from the incorporation of Fe electrolyte impurities within Ni-(oxy)hydroxide, providing direct evidence that a Ni–Fe layered double (oxy)hydroxide (LDH) phase is critical for high OER activity.
Co-reporter:Shannon Klaus
The Journal of Physical Chemistry C 2015 Volume 119(Issue 32) pp:18303-18316
Publication Date(Web):July 19, 2015
DOI:10.1021/acs.jpcc.5b04776
Ni1–xFexOOH thin films prepared via cathodic electrodeposition have been demonstrated to be highly active catalysts for the oxygen evolution reaction (OER) in basic media. Integration of these catalysts with light-absorbing semiconductors is required for photoelectrochemical fuel generation. However, the application of cathodic potentials required for typical electrochemical catalyst deposition limits the library of compatible photoanode materials. Sputter deposition of catalysts circumvents this limitation by enabling facile catalyst layering without cathodic potentials. In this work, we compare the structure and OER activity of sputter-deposited and electrodeposited Ni1–xFexOOH thin films. Electrochemical cycling converts sputtered Ni1–xFex metallic films to the desired oxides/(oxy)hydroxides. Both film preparation methods give catalysts with similar electrochemical behavior across all compositions. Additionally, OER activity is comparable between the deposition methods, with maximum activity for films with ∼20% Fe content (320 mV overpotential at j = 10 mA cm–2 geometric). Electrochemical cycling to convert sputtered metallic Ni1–xFex films to metal oxides/(oxy)hydroxides is found to lower the Fe/Ni ratio, while the electrodeposited films exhibit comparable Fe/Ni ratios before and after electrochemical cycling and characterization. Structurally, Fe is found to incorporate within the Ni(OH)2/NiOOH lattice for films formed through both sputter-deposition and electrodeposition. Layered films were also compared to codeposited 1:1 Fe/Ni films. It is found that, for layered films, an Fe top layer inhibits the electrochemical conversion of metallic Ni to Ni(OH)2/NiOOH, thus reducing the amount of Ni1–xFexOOH OER-active phase formed. In contrast, migration of metals within Ni-on-top films occurs readily during electrochemical cycling, resulting in films that are structurally and electrochemically indistinguishable from codeposited Ni1–xFexOOH. These findings enable direct application of Ni1–xFexOOH sputtered films to a wider library of photoanodes for light-driven water-splitting applications.
Co-reporter:Dr. Sankaranarayanapillai Shylesh;David Hanna;Joseph Gomes;Dr. Christian G. Canlas; Martin Head-Gordon; Alexis T. Bell
ChemSusChem 2015 Volume 8( Issue 3) pp:466-472
Publication Date(Web):
DOI:10.1002/cssc.201402443
Abstract
The catalytic activity of secondary amines supported on mesoporous silica for the self-condensation of n-butanal to 2-ethylhexenal can be altered significantly by controlling the Brønsted acidity of MOH species present on the surface of the support. In this study, MOH (M=Sn, Zr, Ti, and Al) groups were doped onto the surface of SBA-15, a mesoporous silica, prior to grafting secondary propyl amine groups on to the support surface. The catalytic activity was found to depend critically on the synthesis procedure, the nature and amount of metal species introduced and the spatial separation between the acidic sites and amine groups. DFT analysis of the reaction pathway indicates that, for weak Brønsted acid groups, such as SiOH, the rate-limiting step is CC bond formation, whereas for stronger Brønsted acid groups, such as Ti and Al, hydrolysis of iminium species produced upon CC bond formation is the rate-limiting step. Theoretical analysis shows further that the apparent activation energy decreases with increasing Brønsted acidity of the MOH groups, consistent with experimental observation.
Co-reporter:Dr. Sankaranarayanapillai Shylesh;David Hanna;Joseph Gomes;Dr. Christian G. Canlas; Martin Head-Gordon; Alexis T. Bell
ChemSusChem 2015 Volume 8( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/cssc.201400100
Co-reporter:Dr. Eric R. Sacia;Dr. Madhesan Balakrishnan;Matthew H. Deaner;Konstantinos A. Goulas; F. Dean Toste; Alexis T. Bell
ChemSusChem 2015 Volume 8( Issue 10) pp:1726-1736
Publication Date(Web):
DOI:10.1002/cssc.201500002
Abstract
Aviation fuel (i.e., jet fuel) requires a mixture of C9–C16 hydrocarbons having both a high energy density and a low freezing point. While jet fuel is currently produced from petroleum, increasing concern with the release of CO2 into the atmosphere from the combustion of petroleum-based fuels has led to policy changes mandating the inclusion of biomass-based fuels into the fuel pool. Here we report a novel way to produce a mixture of branched cyclohexane derivatives in very high yield (>94 %) that match or exceed many required properties of jet fuel. As starting materials, we use a mixture of n-alkyl methyl ketones and their derivatives obtained from biomass. These synthons are condensed into trimers via base-catalyzed aldol condensation and Michael addition. Hydrodeoxygenation of these products yields mixtures of C12–C21 branched, cyclic alkanes. Using models for predicting the carbon number distribution obtained from a mixture of n-alkyl methyl ketones and for predicting the boiling point distribution of the final mixture of cyclic alkanes, we show that it is possible to define the mixture of synthons that will closely reproduce the distillation curve of traditional jet fuel.
Co-reporter:Dr. Eric R. Sacia;Dr. Madhesan Balakrishnan;Matthew H. Deaner;Konstantinos A. Goulas; F. Dean Toste; Alexis T. Bell
ChemSusChem 2015 Volume 8( Issue 10) pp:
Publication Date(Web):
DOI:10.1002/cssc.201500556
Co-reporter:Dr. Sankaranarayanapillai Shylesh;Daeyoup Kim;Christopher R. Ho;Gregory R. Johnson;Jason Wu; Alexis T. Bell
ChemSusChem 2015 Volume 8( Issue 23) pp:3959-3962
Publication Date(Web):
DOI:10.1002/cssc.201500786
Abstract
Gold nanoparticles (NPs) supported on hydrotalcite (Au/HT) are highly active and selective catalysts for the continuous, gas-phase, non-oxidative dehydrogenation of bioderived C2–C4 alcohols. A sharp increase in turn over frequency (TOF) is noted when the size of Au NPs is less than 5 nm relating to the strong synergy between metallic Au NPs and the acid–base groups on the support surface. It is shown that catalytic activity depends critically on Au NP size, support composition, and support pretreatments. A reaction pathway elucidated from kinetic isotope effects suggests that the abstraction of β-H by Au NPs (C−H activation) is the rate-determining step in the dehydrogenation of bioderived C2–C4 alcohols.
Co-reporter:Dr. Sankaranarayanapillai Shylesh;Daeyoup Kim;Christopher R. Ho;Gregory R. Johnson;Jason Wu; Alexis T. Bell
ChemSusChem 2015 Volume 8( Issue 23) pp:
Publication Date(Web):
DOI:10.1002/cssc.201501451
Co-reporter:Dr. Sankaranarayanapillai Shylesh;Daeyoup Kim;Christopher R. Ho;Gregory R. Johnson;Jason Wu; Alexis T. Bell
ChemSusChem 2015 Volume 8( Issue 23) pp:
Publication Date(Web):
DOI:10.1002/cssc.201501452
Abstract
Invited for this month′s cover is the group of Alexis T. Bell at The University of California, Berkeley in Berkeley, California. The image shows the utility of gold nanoparticles deposited on hydrotalcite (Au/HT) for the continuous gas-phase non- oxidative dehydrogenation reaction of bioderived C2–C4 alcohols to the respective carbonyl compounds and hydrogen. The Communication itself is available at 10.1002/cssc.201500786.
Co-reporter:Amber Janda
The Journal of Physical Chemistry C 2015 Volume 119(Issue 19) pp:10427-10438
Publication Date(Web):April 17, 2015
DOI:10.1021/acs.jpcc.5b01715
Experimental measurements of the rate coefficient (kapp) and apparent enthalpies and entropies of activation (ΔHapp and ΔSapp) for alkane cracking catalyzed by acidic zeolites can be used to characterize the effects of zeolite structure and alkane size on the intrinsic enthalpy and entropy of activation, ΔHint⧧ and ΔSint⧧. To determine ΔHint⧧ and ΔSint⧧, enthalpies and entropies of adsorption, ΔHads-H+ and ΔSads-H+, must be determined for alkane molecules moving from the gas phase to Brønsted acid sites at reaction temperatures (>673 K). Experimental values of ΔHapp and ΔSapp must also be properly defined in terms of ΔHads-H+ and ΔSads-H+. We report here a method for determining ΔHads-H+ and ΔSads-H+ in which the adsorption site is represented by a fixed volume that includes the proton. Values of ΔHads-H+ and ΔSads-H+ obtained from Monte Carlo simulations are in good agreement with values obtained from experimental data measured at 300–400 K. An important feature of the simulations, however, is their ability to account for the redistribution of alkane adsorbed at protons in different locations with increasing temperature. Values of ΔHint⧧ and ΔSint⧧ for the cracking of propane through n-hexane, determined from measured values of kapp and ΔHapp and simulated values of ΔHads-H+ and ΔSads-H+, agree well with values obtained independently from quantum mechanics/molecular mechanics calculations. Application of our method of analysis reveals that the observed increase in kapp with increasing n-alkane size is due primarily to a decrease in ΔHint⧧ with increasing chain length and that ΔSint⧧ is independent of chain length.
Co-reporter:David G. Hanna, Sankaranarayanapillai Shylesh, Yi-Pei Li, Siddarth Krishna, Martin Head-Gordon, and Alexis T. Bell
ACS Catalysis 2014 Volume 4(Issue 9) pp:2908
Publication Date(Web):July 21, 2014
DOI:10.1021/cs500704b
The effects of the coordination environment and connectivity of Ti on the rate of n-butanal self-condensation over Ti-silica catalysts were investigated. Ti was introduced in two ways, either during the synthesis of mesoporous SBA-15 or via grafting onto amorphous silica with a disordered pore structure. The connectivity of Ti was then characterized by XANES, UV–vis, and Raman spectroscopy. For the lowest Ti loadings, the Ti is found to be predominantly in isolated monomeric species, irrespective of the manner of sample preparation, and as the Ti loading is increased, a progressively larger fraction of Ti is present in oligomeric species and anatase nanoparticles. The turnover frequency for butanal condensation decreased monotonically with increasing Ti loading, and the apparent activation energy increased from 60 kJ mol–1 for monomeric species to 120 kJ mol–1 for oligomeric species. A kinetic H/D isotope effect was observed over isolated titanol and Ti dimer catalysts suggesting that α-H abstraction is the rate-determining step. This conclusion is supported by theoretical analysis of the reaction mechanism. In agreement with experimental results, the calculated activation barrier for alkanal condensation over a Ti dimer is roughly two times greater than that over Ti-OH sites. The cause for this difference was explained by energy decomposition analysis of the enolate formation step which showed that there is a large energetic penalty for the substrate to distort over the Ti–O–Ti dimer than the Ti-OH monomer.Keywords: aldol condensation; coordination environment; DFT; kinetics; n-butanal; titanium
Co-reporter:Anton N. Mlinar, Benjamin K. Keitz, David Gygi, Eric D. Bloch, Jeffrey R. Long, and Alexis T. Bell
ACS Catalysis 2014 Volume 4(Issue 3) pp:717
Publication Date(Web):January 27, 2014
DOI:10.1021/cs401189a
Two Ni2+-containing metal–organic frameworks, Ni2(dobdc) and Ni2(dobpdc), are shown to be active for the oligomerization of propene in the gas phase. The metal–organic frameworks exhibit activity comparable to Ni2+-exchanged aluminosilicates but maintain high selectivity for linear oligomers. Thus, these frameworks should enable the high yielding synthesis of linear propene oligomers for use in detergent and diesel fuel applications.Keywords: catalysis; metal−organic frameworks; nickel; oligomerization; propene
Co-reporter:Anton N. Mlinar, Sankaranarayanapillai Shylesh, Otto C. Ho, and Alexis T. Bell
ACS Catalysis 2014 Volume 4(Issue 1) pp:337
Publication Date(Web):December 10, 2013
DOI:10.1021/cs4007809
A series of alkali metal- and nickel-exchanged Al-MCM-41 catalysts were prepared via aqueous ion exchange and then investigated for gas-phase oligomerization of propene at 453 K and near ambient pressures. All catalysts were active and produced oligomers with >98% selectivity. The highest activities per Ni2+ cation were observed when the cations were highly dispersed as a consequence of either lowering the Ni loading for a fixed MCM-41 Si/Al ratio or by decreasing the concentration of exchangeable sites within the material by increasing the MCM-41 Si/Al ratio at a fixed Ni loading. The identity of the alkali metal cation had no significant effect on the catalytic activity or degree of dimer branching, except for the sample containing Cs+ cations, where the decreased pore volume resulted in a lower catalyst activity and slightly more linear dimer products. Comparison of Ni-MCM-41 prepared with and without Na+ cations showed that a higher yield of oligomers could be achieved when Na+ cations are present because of partial removal of strong Brønsted acid sites. For the same reaction conditions, Ni-Na-MCM-41 was more than twice as active as smaller-pored Ni-Na-X zeolites, demonstrating that the activity of Ni2+ cations increases with the increasing free volume near the site. This effect of free volume on the activity of Ni2+ cations was further confirmed by comparing the activities of Ni-Na-X, Ni-Na-MCM-41, Ni-Na-MCM-48, and Ni-Na-SBA-15 with respect to pore size.Keywords: alkali metal; MCM-41; nickel; oligomerization; propene
Co-reporter:Joseph Gomes ; Martin Head-Gordon
The Journal of Physical Chemistry C 2014 Volume 118(Issue 37) pp:21409-21419
Publication Date(Web):August 14, 2014
DOI:10.1021/jp502804q
A hybrid quantum mechanics/molecular mechanics (QM/MM) model and the quasiclassical trajectory (QCT) method have been combined to study the reaction of alkene methylation by methanol catalyzed by the zeolite H-MFI. The rate-limiting step of this reaction is the methylation of the alkene, and the apparent activation energy calculated at the ωB97X-D/6-31G(d,p)//ωB97X-D/6-311++G(3df,3pd) level of theory for this step agrees well with experiment and previous full QM studies. Following the ethene methylation transition state toward the products along the intrinsic reaction coordinate reveals the existence of a protonated cyclopropane (PCP+) carbocation intermediate. A similar protonated methylcyclopropane (mPCP+) carbocation intermediate is found for propene methylation. The intermediates produced during the alkene methylation reaction are metastable with a lifetime of O(1 ps) obtained from QCTs. Because of the short lifetime of these intermediates, the available energy in the carbocation is not in thermal equilibrium distribution with the zeolite lattice before subsequent reaction occurs. The qualitative difference between product distributions obtained by static and dynamic reaction pathways suggests the pathways of zeolite-catalyzed reactions proceed through high-temperature pathways that differ from the 0 K potential energy surface. The transformation of the m-PCP+ intermediate to the longer-lived secondary 2-butyl carbocation observed during QCTs suggests that more stable carbocations can properly thermalize and exist as reaction intermediates for longer than 1 ps.
Co-reporter:Dr. Madhesan Balakrishnan;Eric R. Sacia; Alexis T. Bell
ChemSusChem 2014 Volume 7( Issue 10) pp:2796-2800
Publication Date(Web):
DOI:10.1002/cssc.201402764
Abstract
In this study, we demonstrate that while the energy density and lubricity of the C15 and C16 products of furan condensation of biomass-derived aldehydes with 2-methylfuran are consistent with requirements for diesel, these products do not meet specifications for cetane number and pour point due to their aromatic furan rings. However, a novel class of products that fully meet or exceed most specifications for diesel can be produced by converting the furan rings in these compounds to cyclic ether moieties. Full hydrodeoxygenation of furan condensation products to alkanes would require 55–60 % higher hydrogen demand, starting from biomass, compared to the products of furan ring saturation, providing an additional incentive to support the saturated products. We also report here on a tunable class of catalysts that contain Pd nanoparticles supported on ionic liquid-modified SiO2 that can achieve complete saturation of the furan rings in yields of 95 % without opening these rings.
Co-reporter:Dr. Sankaranarayanapillai Shylesh;David Hanna;Joseph Gomes;Siddarth Krishna;Dr. Christian G. Canlas; Martin Head-Gordon; Alexis T. Bell
ChemCatChem 2014 Volume 6( Issue 5) pp:1283-1290
Publication Date(Web):
DOI:10.1002/cctc.201301087
Abstract
A highly efficient solid-base organocatalyst for the gas-phase aldol self-condensation of n-butanal to 2-ethylhexenal was developed by grafting site-isolated amines on tailored silica surfaces. The catalytic activity depends largely on the nature of amine species, the surface concentration of amine and silanol groups, and the spatial separation between the silanol and amine groups. In situ FTIR measurements demonstrated that the formation of nucleophilic enamines leads to the enhanced catalytic activity of secondary amine catalysts, whereas the formation of imines (stable up to 473 K) leads to the low activity observed for silica-supported primary amines. Blocking the silanol groups on the silica support by silylation or cofeeding water into the reaction stream drastically decreased the reaction rates, demonstrating that weaker acidic silanol groups participate cooperatively with the amine groups to catalyze the condensation reaction. This work demonstrates that the spatial separation of the weakly acidic silanols and amines can be tuned by the controlled dehydration of the supporting silica and by varying the linker length of the amine organosilane precursor used to graft the amine to the support surface. A mechanism for aldol condensation was proposed and then analyzed by DFT calculations. DFT analysis of the reaction pathway suggested that the rate-limiting step in aldol condensation is carboncarbon bond formation, which is consistent with the observed kinetics. The calculated apparent activation barrier agrees reasonably with that measured experimentally.
Co-reporter:Dr. Sebastian Werner;Gregory R. Johnson ; Alexis T. Bell
ChemCatChem 2014 Volume 6( Issue 10) pp:2881-2888
Publication Date(Web):
DOI:10.1002/cctc.201402260
Abstract
Supported Co is an effective catalyst for the Fischer–Tropsch synthesis of various hydrocarbon products that can be converted to diesel. Recent studies have shown that the formation of methane can be suppressed and the formation of C5+ products enhanced by promoting Co with Mn. Because the activity and product selectivity of Co-based catalysts are dependent on the size of Co nanoparticles and the extent of Co promotion by Mn, it is desirable to understand these effects by investigating the performance of Co nanoparticles with well-defined size and elemental composition. The present study was undertaken with the aim of producing well-defined nanoparticles of Co and Co–Mn and then supporting them on silica. Co and Co–Mn particles were synthesized through the polyol reduction of Co and Mn acetylacetonates. By controlling synthesis conditions, Co particles with diameters of 7–10 nm and similarly sized Co–Mn (Mn/Co=0.1) particles were prepared. XRD and elemental mapping with scanning TEM-energy-dispersive X-ray spectroscopy and scanning TEM-electron energy loss spectroscopy studies suggested that most of the Mn species was associated with the Co particles. Ex situ prepared Co and Co–Mn nanoparticles were first supported on silica and then investigated for the catalytic activity for the Fischer–Tropsch synthesis. The turnover frequencies and product distributions obtained with silica-supported Co and Co–Mn nanoparticles were similar to those obtained with catalysts prepared by using the conventional incipient wetness impregnation method. However, the rate of CO consumption per mass of Co was much lower for the catalysts produced by supporting ex situ prepared nanoparticles. This effect was attributed to the sintering of the nanoparticles during their calcination and reduction. Magnetic interactions among nanoparticles during their immobilization and thermal pretreatment were identified as the primary cause of sintering.
Co-reporter:Amber Janda
Journal of the American Chemical Society 2013 Volume 135(Issue 51) pp:19193-19207
Publication Date(Web):November 15, 2013
DOI:10.1021/ja4081937
The aim of this study was to investigate the influence of Si/Al ratio on the locations of exchangeable cations in H-MFI and on the monomolecular cracking and dehydrogenation reactions of n-butane. On the basis of UV–visible spectroscopic analysis of Co(II) exchanged into MFI, it was inferred that the fraction of Co(II) (and, by extension, Brønsted protons) located at channel intersections relative to straight and sinusoidal channels increases with increasing Al content. Concurrently, turnover frequencies for all monomolecular reactions, and the selectivities to dehydrogenation versus cracking and to terminal cracking versus central cracking, generally increased. The changes in selectivity with Al content are consistent with the finding that the transition-state geometry for dehydrogenation is bulky and resembles a product state, and should therefore exhibit a stronger preference to occur at channel intersections relative to cracking. Increases in turnover frequencies are attributed partly to increases in intrinsic activation entropies that compensate for concurrent increases in intrinsic activation energies, most strongly for dehydrogenation and terminal cracking, resulting in increased selectivity to these reactions at higher Al content. This interpretation contrasts with the view that intrinsic activation barriers are constant. It is also observed that isobutene inhibits the rate of n-butane dehydrogenation. Theoretical calculations indicate that this effect originates from adsorption of isobutene at the channel intersections. Because cracking reaction rates are not affected by the presence of isobutene, this result suggests that the preference of dehydrogenation to occur at channel intersections is much stronger than the preference for cracking to occur at these locations.
Co-reporter:Mary W. Louie
Journal of the American Chemical Society 2013 Volume 135(Issue 33) pp:12329-12337
Publication Date(Web):July 16, 2013
DOI:10.1021/ja405351s
A detailed investigation has been carried out of the structure and electrochemical activity of electrodeposited Ni–Fe films for the oxygen evolution reaction (OER) in alkaline electrolytes. Ni–Fe films with a bulk and surface composition of 40% Fe exhibit OER activities that are roughly 2 orders of magnitude higher than that of a freshly deposited Ni film and about 3 orders of magnitude higher than that of an Fe film. The freshly deposited Ni film increases in activity by as much as 20-fold during exposure to the electrolyte (KOH); however, all films containing Fe are stable as deposited. The oxidation of Ni(OH)2 to NiOOH in Ni films occurs at potentials below the onset of the OER. Incorporation of Fe into the film increases the potential at which Ni(OH)2/NiOOH redox occurs and decreases the average oxidation state of Ni in NiOOH. The Tafel slope (40 mV dec–1) and reaction order in OH– (1) for the mixed Ni–Fe films (containing up to 95% Fe) are the same as those for aged Ni films. In situ Raman spectra acquired in 0.1 M KOH at OER potentials show two bands characteristic of NiOOH. The relative intensities of these bands vary with Fe content, indicating a change in the local environment of Ni–O. Similar changes in the relative intensities of the bands and an increase in OER activity are observed when pure Ni films are aged. These observations suggest that the OER is catalyzed by Ni in Ni–Fe films and that the presence of Fe alters the redox properties of Ni, causing a positive shift in the potential at which Ni(OH)2/NiOOH redox occurs, a decrease in the average oxidation state of the Ni sites, and a concurrent increase in the activity of Ni cations for the OER.
Co-reporter:Sankaranarayanapillai Shylesh, David Hanna, Anton Mlinar, Xüé-Qia̅n Kǒng, Jeffrey A. Reimer, and Alexis T. Bell
ACS Catalysis 2013 Volume 3(Issue 3) pp:348
Publication Date(Web):January 15, 2013
DOI:10.1021/cs3007445
An investigation has been carried out to identify the effects of catalyst preparation on the activity, selectivity, and stability of phosphine-modified rhodium/silica catalysts (Rh/SiO2) for propene hydroformylation. High selectivity to aldehydes was achieved, without the formation of propane or butanol. Catalyst activity and selectivity was found to depend strongly on the nature and concentration of the phosphine ligands and the amount of rhodium dispersed on the silica support. Screening of different ligands showed that a bidentate xantphos (X) ligand was ∼2-fold more active than the monodentate phosphine ligand (PPh3) screened at a ligand-to-rhodium ratio of 15:1. Investigation of the effects of reaction temperature, reactant partial pressures, and phosphine-to-rhodium ratio indicates that the kinetics of propene hydroformylation over X-promoted Rh/SiO2 is nearly identical to those for sulfoxantphos-modified rhodium-containing supported ionic liquid phase (SX-Rh SILP) catalysts. In-situ FTIR and solid-state 31P MAS NMR characterization provide evidence for the formation of HRh(CO)n(PPh3)4–n species on PPh3-modified Rh/SiO2, and HRh(CO)2(X) species on xantphos-modified Rh/SiO2. The high catalytic activity observed over rhodium-containing silica catalysts is attributed to formation of Rh(I)(CO)2 species by the process of corrosive chemisorption of Rh nanoparticles by CO and the subsequent ligation of phosphine ligands to the dicarbonyl species. Evidence is also presented suggesting that the active form of the catalyst resides on the surface of the Rh nanoparticles.Keywords: butanal; hydroformylation; phosphine ligands; rhodium; xantphos
Co-reporter:Evan M. W. Rumberger, Hyun S. Ahn, Alexis T. Bell and T. Don Tilley
Dalton Transactions 2013 vol. 42(Issue 34) pp:12238-12247
Publication Date(Web):12 Jul 2013
DOI:10.1039/C3DT51472B
Adsorption of a dinuclear μ-oxo bridged Mn complex onto mesoporous silica was observed when SBA15 was treated with an acetonitrile solution of [Mn2(μ-O)2Cl(μ-O2CCH3)(H2O)(bpy)2](NO3)2 (1). This complex was immobilized via the displacement of NO3− into solution, and characterization by spectroscopic (DRIFTS and DRUV-vis) and magnetic data indicates that the intact dication is electrostatically bound to the silica surface. Loadings of up to 4.1% by weight of [Mn2(μ-O)2Cl(μ-O2CCH3)(H2O)(bpy)2]2+ were achieved. TEM images of the grafted material revealed retention of the mesoporous structure of SBA15, and no clusters of manganese greater than ca. 10 nm were observed. The SBA15-supported dimanganese complex functions as a catalyst for the oxidation of H2O with (NH4)2Ce(NO3)6 as stoichiometric oxidant. In contrast, homogenous aqueous solutions of 1 do not evolve oxygen upon treatment with (NH4)2Ce(NO3)6. Labeling studies with H218O confirm that the oxygen formed in this catalysis is derived from water. Monitoring the O2 evolution allowed determination of an initial rate for the catalysis (TOFi = 1.1 × 10−3 s−1). These studies also reveal a first order dependence on manganese surface concentration, and a zero order rate dependence for (NH4)Ce(NO3)6. Spectroscopic investigations were employed to investigate the difference in activities between dissolved and supported dimanganese complexes.
Co-reporter:Anton N. Mlinar;Otto C. Ho;Gerry G. Bong ; Alexis T. Bell
ChemCatChem 2013 Volume 5( Issue 10) pp:3139-3147
Publication Date(Web):
DOI:10.1002/cctc.201300232
Abstract
A series of alkali metal or alkaline earth-exchanged NiX (X=faujasite zeolite, Si/Al=1.2) zeolites containing approximately 0.6 wt % Ni as Ni2+ cations were examined as catalysts for propene oligomerization at 453 K and 5 bar (500 kPa). In the presence of propene, the activity of alkali metal-exchanged zeolites (NiLiX, NiNaX, and NiKX) increased with time on stream, reached a maximum, then decreased, and finally reached steady state. In contrast, the activity of alkaline earth-exchanged zeolites (NiMgX, NiCaX, and NiSrX) was high initially and then decreased until steady-state activity was achieved. The primary product formed in all cases was hexene (90 %), and nonene was the only other product observed (10 %). Both the rate of propene dimerization and the ratio of branched to linear hexene isomers were determined to depend on the identity of the charge-compensating cation, with the dimerization rate and degree of dimer branching increasing with increasing free volume in the zeolite supercages. The apparent activation energy for trimer formation was greater than that for dimer formation, which suggests that steric constraints imposed by the zeolite may inhibit the growth of larger oligomers.
Co-reporter:Shaama Mallikarjun Sharada, Paul M. Zimmerman, Alexis T. Bell, and Martin Head-Gordon
The Journal of Physical Chemistry C 2013 Volume 117(Issue 24) pp:12600-12611
Publication Date(Web):May 23, 2013
DOI:10.1021/jp402506m
Monomolecular reactions of alkanes in H-MFI were investigated by means of a dispersion-corrected density functional, ωB97X-D, combined with a hybrid quantum mechanics/molecular mechanics (QM/MM) method applied to a cluster model of the zeolite. The cluster contains 437 tetrahedral (T) atoms, within which a T5 region containing the acid site along with the representative alkane is treated quantum mechanically. The influence of active site location on reaction energetics was examined by studying cracking and dehydrogenation reactions of n-butane at two regions in H-MFI–T12, where the proton is at the intersection of straight and sinusoidal channels, and T10, where the proton is within the sinusoidal channel. Two transition states were observed for cracking: one where the proton attacks the C–C bond and another where it attacks a C atom. Dehydrogenation proceeds via a concerted mechanism, where the transition state indicates simultaneous H2 formation and proton migration to the framework. Intrinsic activation energies can be determined accurately with this method, although heats of adsorption were found to be higher in magnitude relative to experiments, which is most likely mainly caused by the MM dispersion parameters for the zeolite framework atoms. Intrinsic activation energies calculated for reactions at the T10 site are higher than those at T12 owing to differences in interaction of the substrate with the acid site as well as with the zeolite framework, demonstrating that Brønsted acid sites in H-MFI are not equivalent for these reactions. Apparent activation energies, determined from calculated intrinsic activation energies and experimentally measured heats of adsorption taken from the literature, are in excellent agreement with experimental results.
Co-reporter:Paul M. Zimmerman ; Diana C. Tranca ; Joseph Gomes ; Daniel S. Lambrecht ; Martin Head-Gordon
Journal of the American Chemical Society 2012 Volume 134(Issue 47) pp:19468-19476
Publication Date(Web):October 15, 2012
DOI:10.1021/ja3089372
Product selectivity of alkane cracking catalysis in the H-MFI zeolite is investigated using both static and dynamic first-principles quantum mechanics/molecular mechanics simulations. These simulations account for the electrostatic- and shape-selective interactions in the zeolite and provide enthalpic barriers that are closely comparable to experiment. Cracking transition states for n-pentane lead to a metastable intermediate (a local minimum with relatively small barriers to escape to deeper minima) where the proton is shared between two hydrocarbon fragments. The zeolite strongly stabilizes these carbocations compared to the gas phase, and the conversion of this intermediate to more stable species determines the product selectivity. Static reaction pathways on the potential energy surface starting from the metastable intermediate include a variety of possible conversions into more stable products. One-picosecond quasiclassical trajectory simulations performed at 773 K indicate that dynamic paths are substantially more diverse than the potential energy paths. Vibrational motion that is dynamically sampled after the cracking transition state causes spilling of the metastable intermediate into a variety of different products. A nearly 10-fold change in the branching ratio between C2/C3 cracking channels is found upon inclusion of post-transition-state dynamics, relative to static electronic structure calculations. Agreement with experiment is improved by the same factor. Because dynamical effects occur soon after passing through the rate-limiting transition state, it is the dynamics, and not only the potential energy barriers, that determine the catalytic selectivity. This study suggests that selectivity in zeolite catalysis is determined by high temperature pathways that differ significantly from 0 K potential surfaces.
Co-reporter:Madhesan Balakrishnan, Eric R. Sacia and Alexis T. Bell
Green Chemistry 2012 vol. 14(Issue 6) pp:1626-1634
Publication Date(Web):19 Mar 2012
DOI:10.1039/C2GC35102A
A low energy intensive process for the production of diesel fuel has been delineated from both 5-(hydroxymethyl)furfural (HMF) and its sugar precursor D-(–)-fructose. Alcoholic solutions of the above produced a mixture of potential bio-diesel candidates namely, 5-(alkoxymethyl)furfural, 5-(alkoxymethyl)furfural dialkylacetal, and alkyl levulinate, in the presence of solid acid catalysts. Sulfonic acid functionalized resins, Amberlyst-15 and Dowex DR2030 showed exceptional reactivity and selectivity for these reactions. Production of another potential diesel candidate 2,5-bis(alkoxymethyl)furan has been optimized through both sequential reduction/etherification and one-pot reductive etherification processes. During the metal catalyzed hydrogenation of HMF, platinum showed an exclusive selectivity for the reduction of the carbonyl functionality of HMF. Both Pt and Pt/Sn supported on Al2O3 catalysts have been optimized for the production of 2,5-bis(alkoxymethyl)furan from HMF. The reaction mechanisms of etherification and reductive etherification have been discussed in detail on the basis of intermediates observed during these processes.
Co-reporter:Sankaranarayanapillai Shylesh, David Hanna, Sebastian Werner, and Alexis T. Bell
ACS Catalysis 2012 Volume 2(Issue 4) pp:487
Publication Date(Web):February 21, 2012
DOI:10.1021/cs2004888
An investigation has been carried out on the effects of catalyst preparation on the activity and stability of supported ionic liquid phase (SILP) catalysts for propene hydroformylation. Catalyst activity and stability were found to be strongly influenced by ligand and ionic liquid composition, ligand-to-rhodium ratio, and the surface density of silanol groups on the silica support. Highest activity was achieved using rhodium sulfoxantphos (SX) complexes in the presence of [bmim][OctSO4]. In situ FT-IR and solid-state 31P and 29Si MAS NMR characterization suggest that active Rh centers are not present as homogeneous complexes dissolved in an ionic liquid film, instead are present as HRh(CO)2SX complexes bound to the support by interactions of the sulfonate groups of SX with silanol groups of the support. The function of the ionic liquid is to inhibit undesired interactions of SX ligands, since such interactions render the phosphine groups unavailable for interaction with the Rh+ cations. Catalyst deactivation is attributed mainly to the formation of catalytically inactive [Rh(CO)(μ-CO)SX]2 or HRh(SX)2 complexes when the SX/Rh ratio is too low or high, respectively.Keywords: hydroformylation; ionic liquids; rhodium; SILP catalysts; sulfoxantphos;
Co-reporter:Zhenmeng Peng, Christian Kisielowski and Alexis T. Bell
Chemical Communications 2012 vol. 48(Issue 13) pp:1854-1856
Publication Date(Web):14 Dec 2011
DOI:10.1039/C2CC16962B
A novel method has been developed for preparing supported cubic platinum nanoparticles. Carbon monoxide and hydrogen are used to reduce platinum precursors present at a solid–gas interface and to control the shape of the growing Pt nanoparticles. By avoiding the use of any organic agents in the synthesis, cubic Pt particles free of hydrocarbons are formed, thereby avoiding possible contamination of the catalyst surface. The approach used is simple and readily scalable.
Co-reporter:Shaama Mallikarjun Sharada, Paul M. Zimmerman, Alexis T. Bell, and Martin Head-Gordon
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 12) pp:5166-5174
Publication Date(Web):October 5, 2012
DOI:10.1021/ct300659d
Accurate and speedy determination of transition structures (TSs) is essential for computational studies on reaction pathways, particularly when the process involves expensive electronic structure calculations. Many search algorithms require a good initial guess of the TS geometry, as well as a Hessian input that possesses a structure consistent with the desired saddle point. Among the double-ended interpolation methods for generation of the guess for the TS, the freezing string method (FSM) is proven to be far less expensive compared to its predecessor, the growing string method (GSM). In this paper, it is demonstrated that the efficiency of this technique can be improved further by replacing the conjugate gradient optimization step (FSM-CG) with a quasi-Newton line search coupled with a BFGS Hessian update (FSM-BFGS). A second crucial factor that affects the speed with which convergence to the TS is achieved is the quality and cost of the Hessian of the energy for the guessed TS. For electronic structure calculations, the cost of calculating an exact Hessian increases more rapidly with system size than the energy and gradient. Therefore, to sidestep calculation of the exact Hessian, an approximate Hessian is constructed, using the tangent direction and local curvature at the TS guess. It is demonstrated that the partitioned-rational function optimization algorithm for locating TSs with this approximate Hessian input performs at least as well as with an exact Hessian input in most test cases. The two techniques, FSM and approximate Hessian construction, therefore can significantly reduce costs associated with finding TSs.
Co-reporter:Ferenc Somodi, Sebastian Werner, Zhenmeng Peng, Andrew Bean Getsoian, Anton N. Mlinar, Boon Siang Yeo, and Alexis T. Bell
Langmuir 2012 Volume 28(Issue 7) pp:3345-3349
Publication Date(Web):February 2, 2012
DOI:10.1021/la204838q
A two-step method has been developed for precise size and composition control of bimetallic Pt–In nanoparticles. Very small (1.62 nm) PtIn seed nanoparticles with 1:1 metal ratio were prepared in the absence of capping agents followed by growth of Pt on their surface in the presence of oleyl amine as reducing and stabilizing agent. Nanoparticles with bulk compositions of Pt4In, Pt3In, and Pt2In could be synthesized with average diameter smaller than 3 nm. TEM, EDX, and XPS provided evidence for homogeneous growth without separate nucleation of pure platinum nanoparticles in the reaction solution. Pt3In nanoparticles were deposited onto SiO2 surface by incipient wetness impregnation. Temperature-induced changes in the particle surface were monitored by in situ IR spectroscopy and CO adsorption. It was found that surface alloy composition of the particles could be tuned by using oxidizing or reducing atmospheres.
Co-reporter:Boon Siang Yeo and Alexis T. Bell
The Journal of Physical Chemistry C 2012 Volume 116(Issue 15) pp:8394-8400
Publication Date(Web):April 11, 2012
DOI:10.1021/jp3007415
Co-reporter:Vladimir Shapovalov, Tim Fievez, and Alexis T. Bell
The Journal of Physical Chemistry C 2012 Volume 116(Issue 35) pp:18728-18735
Publication Date(Web):August 13, 2012
DOI:10.1021/jp302862q
A theoretical model has been developed for describing isolated vanadate species dispersed on the (101) surface of anatase that takes into account the equilibration of the supported species with gas-phase oxygen. The lowest energy of the combined solid and gas phases identifies the VOx species with the optimal structure and composition. This model of VOx species supported on the surface of anatase is then used to analyze the reaction path for methanol oxidation to formaldehyde. The chemisorption of methanol is found to proceed preferentially by addition across a V–O–Ti bond to form V–OCH3 and Ti–OH species. The rate-limiting step for the formation of formaldehyde takes place via the transfer of a hydrogen atom from V–OCH3 bound to an oxygen atom bridging two Ti atoms, i.e., a Ti–O–Ti group located adjacent to the supported vanadate species. This step is found to have the lowest apparent activation energy of all pathways explored for the formation of formaldehyde.
Co-reporter:Andrew Behn, Joseph Zakzeski, Martin Head-Gordon, Alexis T. Bell
Journal of Molecular Catalysis A: Chemical 2012 Volumes 361–362() pp:91-97
Publication Date(Web):September 2012
DOI:10.1016/j.molcata.2012.05.006
The mechanism and kinetics of the liquid-phase, oxidative carbonylation of toluene to toluic acid over Pd(II) in the presence of trifluoroacetic acid (TFAH), trifluoroacetic anhydride (TFAA), and molecular oxygen were investigated through a combination of experimental and theoretical approaches. The experimental results are consistent with the previously proposed mechanism for the oxidative carbonylation of arenes. The reaction is initiated by coordination of toluene to the Pd(II) complex and activation of a CH bond in the benzene ring. This initial step becomes rate limiting when a sufficiently high (NH4VO3)/Pd ratio is used for the reoxidation of Pd(0) to Pd(II). Both processes are found to be dependent on the electron withdrawing and donating capability of the anionic ligands. Overall catalyst activity peaks for ligands of intermediate basicity, and diminishes for both more and less basic ligands. Theoretical analysis of the coordination of toluene and activation of the CH bond on the benzene ring reveals that the basicity of the ligands affects the two processes in opposite ways. Weakly basic ligands promote the coordination of toluene but have the opposite effect on the activation of the CH bond. The tradeoff in these two effects leads to a maximum in the apparent rate coefficient with pKa of the conjugate acid of the anionic ligands. The absence of significant product stereoselectivity is due to a lack of steric hindrance in the binding of toluene to the Pd(II) complex.Graphical abstractHighlights► A combined experimental and theoretical investigation of the oxidative carbonylation of toluene over Pd(II) is performed. ► The mechanism is found to proceed via electrophilic binding toluene and subsequent CH bond activation. ► The role of Pd's ligand structure is investigated, with intermediate acidity ligands producing the most active catalysts. ► The Pd system is compared against previous work with an analogous Rh system.
Co-reporter:Boon Siang Yeo
Journal of the American Chemical Society 2011 Volume 133(Issue 14) pp:5587-5593
Publication Date(Web):March 17, 2011
DOI:10.1021/ja200559j
Scanning electron microscopy, linear sweep voltammetry, chronoamperometry, and in situ surface-enhanced Raman spectroscopy were used to investigate the electrochemical oxygen evolution reaction (OER) occurring on cobalt oxide films deposited on Au and other metal substrates. All experiments were carried out in 0.1 M KOH. A remarkable finding is that the turnover frequency for the OER exhibited by ∼0.4 ML of cobalt oxide deposited on Au is 40 times higher than that of bulk cobalt oxide. The activity of small amounts of cobalt oxide deposited on Pt, Pd, Cu, and Co decreased monotonically in the order Au > Pt > Pd > Cu > Co, paralleling the decreasing electronegativity of the substrate metal. Another notable finding is that the OER turnover frequency for ∼0.4 ML of cobalt oxide deposited on Au is nearly three times higher than that for bulk Ir. Raman spectroscopy revealed that the as-deposited cobalt oxide is present as Co3O4 but undergoes progressive oxidation to CoO(OH) with increasing anodic potential. The higher OER activity of cobalt oxide deposited on Au is attributed to an increase in fraction of the Co sites present as CoIV cations, a state of cobalt believed to be essential for OER to occur. A hypothesis for how CoIV cations contribute to OER is proposed and discussed.
Co-reporter:Sean Dee and Alexis T. Bell
Green Chemistry 2011 vol. 13(Issue 6) pp:1467-1475
Publication Date(Web):27 May 2011
DOI:10.1039/C1GC15317J
Experiments were conducted to study the effects of reaction conditions on the hydrolysis of miscanthus dissolved in 1-ethyl-3-methylimidazolium chloride ([Emim][Cl]) catalyzed by H2SO4. It was determined that while there is a small co-inhibition effect associated with the simultaneous hydrolysis of the cellulosic and hemicellulosic portions of miscanthus, the largest rate decreases were observed for the hydrolysis of the hemicellulosic portion. This rate decrease was attributed to the chemical linkage between hemicellulose and lignin in miscanthus, which could be broken with chemical pretreatment. While chemical pretreatment increased the rate of the hydrolysis of the hemicellulosic component, delignification showed no further benefit. The rate of hydrolysis was determined to be first order in concentrations of β-1,4 glycosidic linkage and acid, and zero order in water concentration. The activation energy for the hydrolysis of the glycosidic linkages in the cellulosic and hemicellulosic components were determined to be 95 kJ mol−1 and 114 kJ mol−1 respectively. Progressive addition of water during the first hour of the reaction increased conversion and selectivity to saccharine products, while limiting dehydration of the sugars formed. The conversion of the cellulosic portion of miscanthus could be increased after the first hour of the reaction by increasing the reactor temperature. While miscanthus is only partially soluble in [Emim][Cl], it was found that the initial miscanthus loading could be increased to 9 wt% before significant yield decreases attributed to solubility limitations of the cellulosic component were observed. By proper adjustment of reaction conditions, it was possible to achieve yields of sugars approaching 84% from the cellulosic and hemicellulosic components of miscanthus, with minimal dehydration of the sugars to furans.
Co-reporter:Suzanne Lutfalla, Vladimir Shapovalov, and Alexis T. Bell
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 7) pp:2218-2223
Publication Date(Web):June 7, 2011
DOI:10.1021/ct200202g
GGA+U calculation were performed for oxides of Ti, V, Mo, and Ce with the objective of establishing the best value of the parameter Ueff to use in order to match the calculated reduction and oxidation energies of each oxide with experimental values. In each case, the reaction involved the hydrogen reduction of an oxide to its next lower oxide and the formation of water. Our calculations show that the optimal value of Ueff required to match calculated and experimental values of the reaction energy are significantly different from those reported in the literature based on matching lattice parameters or electronic properties and that the use of these values of Ueff can result in errors in the calculated redox energies of over 100 kJ/mol. We also found that, when an element exhibits more than two oxidation states, the energy of redox reactions between different pairs of these states are described by slightly different values of Ueff.
Co-reporter:Sean J. Dee ; Alexis T. Bell
ChemSusChem 2011 Volume 4( Issue 8) pp:1166-1173
Publication Date(Web):
DOI:10.1002/cssc.201000426
Abstract
An investigation was carried out into the hydrolysis of cellulose dissolved in 1-ethyl-3-methylimidazolium chloride ([Emim][Cl]) and 1-butyl-3-methylimidazolium chloride ([Bmim][Cl]) catalyzed by mineral acids. Glucose, cellobiose, and 5-hydroxymethylfurfural (5-HMF) were observed as the primary reaction products. The initial rate of glucose formation was determined to be of first order in the concentrations of dissolved glucan and protons and of zero order in the concentration of water. The absence of a dependence on water concentration suggests that cleavage of the β-1,4-glycosidic linkages near chain ends is irreversible. The apparent activation energy for glucose formation is 96 kJ mol−1. The absence of oligosaccharides longer than cellobiose suggests that cleavage of interior glycosidic bonds is reversible due to the slow diffusional separation of cleaved chains in the highly viscous glucan/ionic liquid solution. Progressive addition of water during the course of glucan hydrolysis inhibited the rate of glucose dehydration to 5-HMF and the formation of humins. The inhibition of glucose dehydration is attributed to stronger interaction of protons with water than the 2-OH atom of the pyranose ring of glucose, the critical step in the proposed mechanism for the formation of 5-HMF. The reduction in humin formation associated with water addition is ascribed to the lowered concentration of 5-HMF, since the formation of humins is suggested to proceed through the condensation polymerization of 5-HMF with glucose.
Co-reporter:Taejin Kim;Fuat E. Celik;David G. Hanna;S. Shylesh
Topics in Catalysis 2011 Volume 54( Issue 5-7) pp:299-307
Publication Date(Web):2011 April
DOI:10.1007/s11244-011-9664-3
Heterogeneous rhodium catalysts supported on SiO2 were modified with PPh3 for the gas-phase hydroformylation of propene to produce n- and isobutanal. High selectivity to aldehydes was achieved, with no propane or alcohols formed. Investigation of the effects of reaction temperature, reactant partial pressures, total pressure, and PPh3/Rh ratio suggested that the supported catalyst behaved similarly to the homogeneous catalyst. In particular, the supported catalyst showed similar activation energies and partial and total pressure dependences of the reaction rates to those observed in homogeneous, liquid-phase reactions. The first order dependence of the hydroformylation rate on the partial pressures of propene, CO, and H2 individually led to a cubic dependence of butanal formation on total pressure for equimolar reactant mixtures. High regioselectivity with a typical n/i ratio of 14 was achieved.
Co-reporter:Ferenc Somodi ; Zhenmeng Peng ; Andrew “Bean” Getsoian
The Journal of Physical Chemistry C 2011 Volume 115(Issue 39) pp:19084-19090
Publication Date(Web):August 23, 2011
DOI:10.1021/jp206482y
Platinum–tin alloy nanoparticles with cubic and hexagonal structure have been prepared by the “heating up” method using metal–acetylacetonates as precursors and 1,2-hexadecanediol as the reducing agent dissolved in dioctyl ether containing oleyl amine and oleic acid as capping agents. The influence of the principal reaction parameters—the concentration of the capping agents, final reaction temperature, reaction time, and ratio of metal precursors—on the size and composition of the Pt–Sn nanoparticles was investigated. Transmission electron microscopy and X-ray diffraction results show that decreasing the amount of capping agents increases not only the size of the nanoparticles but also the extent of alloying. It is proposed that the reaction between the metal precursors is the primary step of the nucleation process leading to Pt–Sn bimetallic particles. In a competitive reaction that depends on the concentration of capping agents, metal oleate complexes are formed. The balance between the rates of these processes affects the relative rates of particle nucleation and growth as well as the composition of the bimetallic nanoparticles. The preparation method described is suitable for controlled formation of Pt–Sn nanoparticles with cubic and hexagonal crystalline structure, which are excellent candidates for investigation of the structure–activity relationship for a number of catalyzed reactions.
Co-reporter:Mandan Chidambaram and Alexis T. Bell
Green Chemistry 2010 vol. 12(Issue 7) pp:1253-1262
Publication Date(Web):28 May 2010
DOI:10.1039/C004343E
Lignocellulosic biomass is an attractive resource for producing transportation fuels, and consequently novel approaches are being sought for transforming the lignin and cellulosic constituents of biomass to fuels or fuel additives. Glucose, the monomer of cellulose, is a good starting material for exploring such chemistries. We report here the results of an investigation aimed at identifying catalysts for the dehydration of glucose to 5-hydroxymethylfurfural (HMF) dissolved in ionic liquids and the subsequent conversion of HMF to 2,5-dimethylfuran (DMF), a high-energy content product that could be used as a fuel or fuel additive. Heteropoly acids were found to be exceptionally active and selective catalysts for the dehydration of glucose. Nearly 100% yield of HMF could be achieved using 12-molybdophosphoric acid (12-MPA) in a solution of 1-ethyl-3-methylimidazolium chloride (EMIMCl) and acetonitrile. The addition of acetonitrile to EMIMCl suppressed the formation of humins from glucose. The high HMF selectivity achievable with heteropoly acid catalysts is ascribed to stabilization of 1,2-enediol and other intermediates involved in the dehydration of glucose and the avoidance of forming the 2,3-enediol intermediate leading to furylhydroxymethyl ketone (FHMK). Carbon-supported metals, and in particular Pd/C, were effective in promoting the hydrogenation of HMF dissolved in EMIMCl and acetonitrile to DMF. The following intermediates were observed in the hydrogenation of HMF to DMF: 5-methylfurfural (MF), 5-methylfurfyl alcohol (MFA), and 2,5-dihydroxymethylfuran (DHMF). The relative rate of formation and consumption of these compounds was explored by using each of them as a reactant in order to identify the reaction pathway from HMF to DMF. It was also observed that HMF produced via glucose dehydration could be converted to DMF without isolation, if the dehydration catalyst, 12 MPA, was replaced by the hydrogenation catalyst, Pd/C. This two-step catalytic approach provides the basis for completely converting glucose to HMF and further converting HMF to DMF.
Co-reporter:Andrew Behn, Martin Head-Gordon and Alexis T. Bell
Organometallics 2010 Volume 29(Issue 5) pp:1144-1149
Publication Date(Web):February 11, 2010
DOI:10.1021/om900945z
A theoretical study was conducted to investigate the chemical nature of an unusual interaction observed between carbonyl and acetate ligands in the Rh(CO)2(CF3COO)3 complex. This interaction is intriguing because it is only nominally longer (0.1−0.2 Å) than a typical carbon−oxygen σ bond, yet is associated with only a modest (∼10 kcal/mol) energy lowering of the complex. A localized bonding molecular orbital that promotes the notion of charge sharing is present between the interacting ligands. Constrained geometry optimizations in tandem with Mulliken population analyses indicate that the interaction stems from the inability of Rh(III) with highly electron withdrawing ligands to back-donate properly into the carbonyl ligands. This produces a charge imbalance in the ligands, which sets the stage for nucleophilic attack by the acetate oxygen to the carbonyl carbon. This interaction causes a shift in the predicted values of both IR and 13C NMR signals, which are compared to experiment. For a full explanation of the 13C NMR shifts, two explicit solvent molecules were added to the model and found to induce interaction of both carbonyls with acetate ligands. The chosen density functional (B3LYP) and basis set were validated by comparing theoretically predicted structures and vibrational frequencies with experimentally determined values for several complexes.
Co-reporter:Ting Chen, Mandan Chidambaram, Zhiping Liu, Berend Smit and Alexis T. Bell
The Journal of Physical Chemistry B 2010 Volume 114(Issue 17) pp:5790-5794
Publication Date(Web):April 13, 2010
DOI:10.1021/jp911372j
A recently improved ionic liquid force field was used to compute the viscosity for binary and ternary mixtures of 1-ethyl-3-methylimidazolium chloride ([emim][Cl]) with water, acetonitrile, and glucose. For the same systems, experimental viscosity data are provided. The simulation and experimental results were in reasonable agreement. Simulations consistently overestimate the viscosities for the mixtures of [emim][Cl] and glucose while the viscosities of the mixtures of glucose and water are well reproduced. Both experiments and simulations show that the addition of acetonitrile reduces the viscosity of a solution of [emim][Cl] and glucose by more than an order of magnitude.
Co-reporter:Jennifer Strunk ; William C. Vining
The Journal of Physical Chemistry C 2010 Volume 114(Issue 40) pp:16937-16945
Publication Date(Web):June 11, 2010
DOI:10.1021/jp100104d
The reduction and reoxidation of submonolayer coverages of TiO2 deposited onto MCM-48 were investigated. The deposited TiO2 was characterized by Raman and UV−visible spectroscopy. Raman spectra show that Ti atoms are bonded to the silica support by Ti−O−Si bonds and that crystalline TiO2 is not formed. The results of the Raman and UV−visible spectroscopy suggest that the dispersed TiO2 is present as two-dimensional oligomeric structures. Reduction in H2 at 923 K produces Ti3+ cations observable by EPR (g = 1.932), suggesting the formation of oxygen vacancies. The fraction of Ti that could be reduced increased with TiO2 surface concentration. This observation is attributed to the ease with which O atoms can be removed from the TiO2 overlayer as the size of the titania patches increases. The amount of oxygen removed during reduction was quantified by pulsed reoxidation. It was observed that the temperature required for complete reoxidation decreased with increasing surface coverage of the silica support by TiO2. This trend is explained with a proposed model of the reoxidation process, in which the rate limiting step is the migration of peroxide species through or between the deposited TiO2 patches. A linear correlation was established between the intensity of the EPR signal for Ti3+ and the amount of oxygen removed from TiO2/SiO2. This relationship was then used to determine the oxygen vacancy concentration present on the surface of TiO2/SiO2 after temperature-programmed oxidation of methanol.
Co-reporter:Joseph A. Swisher, Niels Hansen, Theo Maesen, Frerich J. Keil, Berend Smit and Alexis T. Bell
The Journal of Physical Chemistry C 2010 Volume 114(Issue 22) pp:10229-10239
Publication Date(Web):May 14, 2010
DOI:10.1021/jp101262y
The kinetics of alkane cracking in zeolites MFI and FAU have been simulated theoretically from first principles. The apparent rate coefficient for alkane cracking was described as the product of the number of alkane molecules per unit mass of zeolite that are close enough to a Brønsted-acid site to be in the reactant state for the cleavage of a specific C−C bond and the intrinsic rate coefficient for the cleavage of that bond. Adsorption thermodynamics were calculated by Monte Carlo simulation and the intrinsic rate coefficient for alkane cracking was determined from density functional theory calculations combined with absolute rate theory. The effects of functional, basis set, and cluster size on the intrinsic activation energy for alkane cracking were investigated. The dependence of the apparent rate coefficient on the carbon number for the cracking of C3−C6 alkanes on MFI and FAU determined by simulation agrees well with experimental observation, but the absolute values of the apparent rate coefficients are a factor of 10 to 100 smaller than those observed. This discrepancy is attributed to the use of a small T5 cluster representation of the Brønsted-acid site. Limited calculations for propane and butane cracking on MFI reveal that significantly better agreement between prediction and observation is achieved using a T23 cluster for both the apparent rate coefficient and the apparent activation energy. The apparent rate coefficients for alkane cracking are noticeably larger for MFI than FAU, in agreement with recent findings reported in the experimental literature.
Co-reporter:Boon Siang Yeo Dr.;Shannon L. Klaus;Philip N. Ross Dr.;Richard A. Mathies
ChemPhysChem 2010 Volume 11( Issue 9) pp:1854-1857
Publication Date(Web):
DOI:10.1002/cphc.201000294
Co-reporter:Arne Dinse, Reinhard Schomäcker and Alexis T. Bell
Physical Chemistry Chemical Physics 2009 vol. 11(Issue 29) pp:6119-6124
Publication Date(Web):11 May 2009
DOI:10.1039/B821131K
The oxidative dehydrogenation (ODH) of ethane on alumina-supported vanadia was investigated with the aim of understanding the effects of lattice oxygen and vanadium oxidation state on the catalyst ODH activity and ethene selectivity. Transient-response experiments were carried out with both a fully oxidized sample of 10 wt% VOx/Al2O3 (7 V nm−2) and a sample that had been partially reduced in H2. The experimental results were analyzed to determine the rate coefficients for ethane ODH, k1, and ethene combustion, k3. The rate of ODH was found to depend solely on the concentration of reactive oxygen in the catalyst, but not on the means by which this oxygen concentration was attained (i.e., by H2versus C2H6 reduction). On the other hand, the ethene selectivity observed at a given concentration of active oxygen was found to depend on the composition of the reducing agent, higher ethene selectivities being observed when H2, rather than C2H6, was used as the reducing agent. It is proposed that the higher ethene selectivity achieved by H2versus C2H6 reduction might be due to a lower ratio of V4+ to V3+ cations attained upon reduction in H2 for a given extent of V5+ reduction. This interpretation is based on the hypothesis that ethene combustion is initiated by C2H4 adsorption on Vn+ cations present at the catalyst surface and that the strength of adsorption decreases in the order V5+ > V4+ > V3+ consistent with the decreasing Lewis acidity of the cations.
Co-reporter:Joseph Zakzeski, Sarah Burton, Andrew Behn, Martin Head-Gordon and Alexis T. Bell
Physical Chemistry Chemical Physics 2009 vol. 11(Issue 42) pp:9903-9911
Publication Date(Web):21 Aug 2009
DOI:10.1039/B906883J
A spectroscopic investigation of complexes used to catalyze the oxidative carbonylation of toluene to p-toluic acid was conducted. Rhodium complexes were analyzed by 103Rh and 13C NMR, UV-visible spectroscopy, and infrared spectroscopy. In the presence of vanadium and oxygen, the resting state of the Rh-catalyst was found to exist as a Rh(III) complex with carbonyl and trifluoroacetate ligands, consistent with the structure Rh(CO)2(TFA)3. The 13C NMR spectrum of Rh(13CO)2(TFA)3 complex exhibited a carbonyl peak with an unusual degree of shielding, which resulted in the appearance of the carbonyl peak at an unprecedented upfield position in the 13C NMR spectrum. This shielding was caused by interaction of the carbonyl group with the trifluoroacetate ligand. In the absence of oxygen, the Rh(III) complex reduced to Rh(I), and the reduced form exhibited properties resembling the catalyst precursor. Structures and spectroscopic properties calculated using density functional theory agreed closely with the experimental results. The vanadium co-catalyst used to reoxidize Rh(I) to Rh(III) was similarly characterized by 51V NMR and UV-visible spectroscopy. The oxidized species corresponded to [(VO2)(TFA)]2, whereas the reduced species corresponded to (VO)(TFA)2. The spectroscopic results obtained in this study confirm the identity of the species that have been proposed to be involved in the Rh-catalyzed oxidative carbonylation of toluene to toluic acid.
Co-reporter:FuatE. Celik;Tae-Jin Kim Dr. ;AlexisT. Bell
Angewandte Chemie International Edition 2009 Volume 48( Issue 26) pp:4813-4815
Publication Date(Web):
DOI:10.1002/anie.200900464
Co-reporter:Michael Zboray, Alexis T. Bell and Enrique Iglesia
The Journal of Physical Chemistry C 2009 Volume 113(Issue 28) pp:12380-12386
Publication Date(Web):June 19, 2009
DOI:10.1021/jp901595k
The oxidative dehydrogenation of alkanes (C2H6, C3H8, i-C4H10, and n-C4H10) was investigated on VOx supported on Al2O3. Rate constants for alkane dehydrogenation (k1), alkane combustion (k2), and alkene combustion (k3) were measured, and a model was developed to describe the effects of alkane composition on these rate constants. The proposed model accounts for the effects of the number of C−H bonds available for activation and the relative strengths of these bonds in both the reactant and the product molecules. The Brønsted−Evans−Polanyi (BEP) relationship is used to relate activation energies of secondary and tertiary C−H bonds to that of primary C−H bonds. The model gives a reasonable approximation of the relative order of alkane reactivity, expressed by k1 + k2, and the relative ranking of alkanes with respct to combustion versus oxidative dehydrogenation, expressed by k2/k1. The ratio of k2/k1 is described by the product of two components: one that depends on the nature, number, and relative strength of C−H bonds of surface alkoxides, and a second one that is independent of the alkoxide composition and structure but depends on the difference in the entropy of activation for COx precursor versus alkene formation. The model also explains the observed variation of k3 with alkene composition by considering two precursor states for alkenes. One is strongly bound through π-orbital interactions with Lewis acid centers, and the second weakly binds via H bonding and van der Waals interactions, similar to the binding of alkanes. As a result, the rate of alkene combustion depends strongly on the large heats of adsorption of alkenes and only slightly on the presence of weak allylic C−H bonds. The high rate of C2H4 combustion is thus a consequence of its high heat of adsorption.
Co-reporter:Joseph Zakzeski, Alexis T. Bell
Journal of Molecular Catalysis A: Chemical 2009 Volume 302(1–2) pp:59-67
Publication Date(Web):1 April 2009
DOI:10.1016/j.molcata.2008.11.034
The oxidative carbonylation of benzotrifluoride to form trifluoromethylbenzoic acid (TFMBA) has been catalyzed using either Rh(III) or a Pd(II) cation in combination with a carboxylic acid and its anhydride, ammonium metavanadate, CO, and O2. The influence of metal cation and vanadate concentrations, temperature, time, acid composition, and gas pressures, were explored. The accumulated data suggest that oxidative carbonylation of benzotrifluoride proceeds via an electrophilic mechanism. Rh-catalyzed reactions exhibited higher activity (∼70 turnovers in 4 h at 353 K) and trifluoromethylbenzoic acid selectivity (∼92–95%) than Pd-catalyzed reactions (∼6 turnovers in 4 h at 353 K and a trifluoromethylbenzoic acid selectivity of 80%). Formation of the principle reaction byproducts, benzoyl fluoride and benzoic acid, was highly dependant on the V(V) concentration and the acid composition. Reactions conducted in the presence of CF3COOH yielded the highest catalytic activity, but reactions conducted in CCl3COOH resulted in the highest trifluoromethylbenzoic acid selectivity. A possible mechanism for the reaction has been proposed.The oxidative carbonylation of benzotrifluoride to form trifluoromethylbenzoic acid (TFMBA) has been catalyzed using either Rh(III) or a Pd(II) cation in combination with a carboxylic acid and its anhydride, ammonium metavanadate, CO, and O2. The influence of metal cation and vanadate concentrations, temperature, time, acid composition, and gas pressures, were explored.
Co-reporter:Arthur J. Esswein, Meredith J. McMurdo, Phillip N. Ross, Alexis T. Bell and T. Don Tilley
The Journal of Physical Chemistry C 2009 Volume 113(Issue 33) pp:15068-15072
Publication Date(Web):July 28, 2009
DOI:10.1021/jp904022e
Cubic Co3O4 nanoparticles with average diameters of 5.9, 21.1, and 46.9 nm (hereafter small, medium, and large) have been synthesized and characterized by pXRD, TEM, and BET. The nanoparticles were loaded onto Ni foam supports for evaluation as anodes for water electrolysis in 1.0 M KOH. Current densities of 10 mA/cm2 were achieved at overpotentials of 328, 363, and 382 mV for anodes loaded with 1 mg/cm2 of small, medium, and large sized Co3O4 nanoparticles, respectively. The activity correlates with the BET surface area of the isolated particles. A plot of the electrochemical overpotential at 10 mA/cm2 against the log of the BET surface area gives a linear relation with a slope of −47 ± 7 mV/dec, showing unequivocally that the activity increase is a function of accessible catalyst surface area.
Co-reporter:FuatE. Celik;Tae-Jin Kim Dr. ;AlexisT. Bell
Angewandte Chemie 2009 Volume 121( Issue 26) pp:4907-4909
Publication Date(Web):
DOI:10.1002/ange.200900464
Co-reporter:Niels Hansen ; Till Brüggemann ; Alexis T. Bell ;Frerich J. Keil
The Journal of Physical Chemistry C 2008 Volume 112(Issue 39) pp:15402-15411
Publication Date(Web):September 10, 2008
DOI:10.1021/jp8036022
Benzene alkylation with ethene over zeolite H-ZSM-5 has been investigated using density functional theory. Three different reaction mechanisms—two one-step schemes and one two-step scheme—have been studied on three cluster models of increasing size representing parts of the H-ZSM-5 framework. In the one-step schemes ethene protonation and C−C bond formation occur simultaneously. The two-step scheme starts with the formation of a stable ethoxide intermediate which subsequently reacts with benzene to form the reaction product. Activation energies obtained from the DFT results have been improved by single-point MP2 calculations. The calculated intrinsic activation energies of the one-step schemes are similar to the activation energy of the alkylation step in the two-step scheme. Numerical values of the MP2 corrected activation energies are in good agreement with experimental data. The largest cluster (33 T-atoms) was found to stabilize protonated ethylbenzene as a stable intermediate. The results of this study show the importance of using relatively large clusters for investigations of hydrocarbon transformation occurring in zeolites.
Co-reporter:Sudip Mukhopadhyay;Mark Zerella;Alexis T. Bell
Advanced Synthesis & Catalysis 2005 Volume 347(Issue 9) pp:
Publication Date(Web):19 JUL 2005
DOI:10.1002/adsc.200404394
A direct approach for producing methanol from methane in a three-step, liquid phase process is reported. In the first step, methane is reacted with SO3 to form methanesulfonic acid (MSA) at 75 °C using a free-radical initiator and MSA as the solvent. Urea-H2O2 in combination with RhCl3 is found to be the most effective initiator (57% conversion of SO3; 7.2% conversion of CH4). MSA is then oxidized by SO3 at 160 °C in a second step to produce a mixture containing methyl bisulfate and some methyl methanesulfonate (87% conversion of MSA). In the third step, the mixture of methyl bisulfate and methyl methanesulfonate is hydrolyzed in the presence of an organic solvent, to produce an organic phase containing methanol and an aqueous phase containing sulfuric acid and some MSA (63% conversion of methyl bisulfate; 72% conversion of methyl methanesulfonate). Overall, 58% of the MSA (of which 23% is derived from methane) is converted to methanol.
Co-reporter:Sudip Mukhopadhyay, Mark Zerella, Alexis T. Bell, R. Vijay Srinivas and Gary S. Smith
Chemical Communications 2004 (Issue 4) pp:472-473
Publication Date(Web):27 Jan 2004
DOI:10.1039/B314160H
Methane is transformed selectively to methanesulfonyl chloride at low temperature by liquid-phase reaction of methane with SO2Cl2 in the presence of a free radical initiator and a promoter using 100% H2SO4 as the solvent.
Co-reporter:Mark Zerella, Sudip Mukhopadhyay and Alexis T. Bell
Chemical Communications 2004 (Issue 17) pp:1948-1949
Publication Date(Web):03 Aug 2004
DOI:10.1039/B405549G
Methane is catalytically converted primarily to acetic acid in concentrated sulfuric acid using a combination of Pd2+ and Cu2+ in the presence of oxygen.
Co-reporter:Sudip Mukhopadhyay;Alexis T. Bell
Advanced Synthesis & Catalysis 2004 Volume 346(Issue 8) pp:
Publication Date(Web):5 AUG 2004
DOI:10.1002/adsc.200404060
Direct sulfonation of methane with SO3 to methanesulfonic acid (MSA) is accomplished in sulfuric acid with almost 100% selectivity in the presence of a catalyst, namely, Ce and Rh salts and molecular oxygen as the catalyst regenerator. In the absence of O2, the catalyst remains effective but the selectivity to MSA decreases to 53% and byproducts, principally CH3OSO3H, are formed. The effects of O2 pressure, catalyst concentration, temperature, SO3 concentration, and methane pressure have been examined on the rate of SO3 conversion to MSA. The conversion of SO3 to MSA was the same when CF3SO3H was used as the solvent instead of H2SO4.
Co-reporter:Morris D. Argyle, Kaidong Chen, Carlo Resini, Catherine Krebs, Alexis T. Bell and Enrique Iglesia
Chemical Communications 2003 (Issue 16) pp:2082-2083
Publication Date(Web):15 Jul 2003
DOI:10.1039/B305264H
The extent of reduction of active centers during oxidative alkane dehydrogenation on VOx/Al2O3 was measured from pre-edge UV-visible spectral features and found to increase with increasing VOx domain size and propane/O2 ratio.
Co-reporter:Sudip Mukhopadhyay and Alexis T. Bell
Chemical Communications 2003 (Issue 13) pp:1590-1591
Publication Date(Web):03 Jun 2003
DOI:10.1039/B303561A
Methane is transformed selectively to methanesulfonic acid at low temperature by liquid-phase sulfonation of methane with SO2 and O2 in the presence of Pd- and Cu-salts as the catalysts.
Co-reporter:Sudip Mukhopadhyay
Angewandte Chemie 2003 Volume 115(Issue 26) pp:
Publication Date(Web):2 JUL 2003
DOI:10.1002/ange.200350976
Bei niedrigen Temperaturen und Drücken gelingt die Sulfonierung von Methan mit einem geeigneten Katalysator und Radikalstarter [Gl. (1); MSA=Methansulfonsäure]. Die Weiterentwicklung dieser Reaktion zum industriellen Prozess erscheint vielversprechend: Der Initiator-Komplex ist sowohl stabil, als auch leicht zu handhaben, und der RhCl3-Katalysator kann zurückgewonnen werden.
Co-reporter:Sudip Mukhopadhyay Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 9) pp:
Publication Date(Web):26 FEB 2003
DOI:10.1002/anie.200390260
Co-reporter:Sudip Mukhopadhyay
Angewandte Chemie International Edition 2003 Volume 42(Issue 26) pp:
Publication Date(Web):2 JUL 2003
DOI:10.1002/anie.200350976
Low temperatures and low pressures suffice for the sulfonation of methane with a suitable free-radical initiator and promoter [Eq. (1)]. Since the RhCl3 promoter can be recycled, and the initiator complex is stable and easy to handle, development of this reaction into an industrial process is promising. MSA=methanesulfonic acid.
Co-reporter:Sudip Mukhopadhyay Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 26) pp:
Publication Date(Web):2 JUL 2003
DOI:10.1002/anie.200390508
Co-reporter:Sudip Mukhopadhyay Dr.
Angewandte Chemie 2003 Volume 115(Issue 26) pp:
Publication Date(Web):2 JUL 2003
DOI:10.1002/ange.200390537
Co-reporter:Eric R. Sacia, Madhesan Balakrishnan, Alexis T. Bell
Journal of Catalysis (May 2014) Volume 313() pp:70-79
Publication Date(Web):1 May 2014
DOI:10.1016/j.jcat.2014.02.012
•Biodiesel is produced by etherification of methylfurfural alcohol (MFA) with ethanol or butanol.•Amberlyst 15 catalyzes the selective formation of alkyl-furanyl ethers.•The kinetics of MFA etherification is first order in MFA and catalyst.•Electron-donating/withdrawing substituents affect the rate of furanyl alcohol etherification.The etherification of furanyl alcohols produced from biomass-derived glucose and fructose has been a growing area of research for production of alternative diesel additives. We have determined that the Brønsted acidic resin catalyst, Amberlyst-15, is highly active and selective for the etherification of furanyl alcohols by both ethanol and butanol. The mechanism and kinetics of this reaction were investigated using 5-methylfurfuryl alcohol (MFA) as a probe molecule. Etherification of MFA was found to be first order in both the concentrations of furanyl alcohol and the acid sites. The mechanism of MFA etherification also holds for the etherification of 2,5-bis(hydroxymethyl)furan (BHMF) and 5-(hydroxymethyl)furfural (HMF). In the case of HMF, we find that acetalization of HMF precedes etherification in alcohol solutions. The apparent activation energy of furanyl alcohol etherification in ethanol and butanol solutions ranged from 17.0 to 26.3 kcal/mol. Electron donation/withdrawal at the 2 or 5 position of the furan ring in addition to solvent polarity was found to have significant effects on the rate of furanyl alcohol etherification.Graphical abstractDownload high-res image (80KB)Download full-size image
Co-reporter:Jason Wu, Zhenmeng Peng, Alexis T. Bell
Journal of Catalysis (March 2014) Volume 311() pp:161-168
Publication Date(Web):1 March 2014
DOI:10.1016/j.jcat.2013.11.017
•PtxSn100−x/Mg(Al)O (70 ⩽ x ⩽ 100) catalysts are prepared with controlled particle size and composition.•The specific activity of PtxSn100−x/Mg(Al)O for ethane dehydrogenation increases with increasing metal particle size and increasing Sn/Pt ratio.•Carbon deposition decreases with decreasing metal particle size and increasing Sn/Pt ratio.The effects of composition and metal particle size of platinum catalysts on ethane dehydrogenation were investigated on PtxSn100−x/Mg(Al)O (70 ⩽ x ⩽ 100) catalysts prepared with average particle sizes between ∼2 and 7 nm. At high conversions, catalyst deactivation from coke formation was a strong function of particle size and Sn/Pt. Deactivation decreased significantly with decreasing particle size and increasing Sn addition. To understand the true initial activity of un-deactivated catalysts, further experiments were run at low residence time conditions to limit ethene formation. For a fixed average particle size, ethane TOF and the selectivity to ethene increased with increasing content of Sn in the PtSn particle. For Pt and Pt3Sn compositions, ethane TOF increased with increasing particle size, while the selectivity to ethene was not strongly affected. The observed effects of particle size and composition are attributed to a combination of geometric and electronic factors.The effects of composition and metal particle size of platinum catalysts on ethane dehydrogenation were investigated by studying PtxSn100−x/Mg(Al)O (70 ⩽ x ⩽ 100) catalysts with a good control over both parameters, which exhibit strong size and composition dependency in terms of the TOF and selectivity of ethene production as well as the accumulation of coke.Download high-res image (62KB)Download full-size image
Co-reporter:David G. Hanna, Sankaranarayanapillai Shylesh, Pedro A. Parada, Alexis T. Bell
Journal of Catalysis (March 2014) Volume 311() pp:52-58
Publication Date(Web):1 March 2014
DOI:10.1016/j.jcat.2013.11.012
•Shvo/SiO2 is active for the hydrogenation of butanal in the presence of CO.•Butanal hydrogenation is rate-limited by butanal coordination/reduction.•A proposed mechanism describes the observed kinetics of butanal hydrogenation.•Propene hydroformylation and subsequent butanal hydrogenation produces butanol.The objective of the present study was to develop a heterogeneous catalyst for the hydrogenation of butanal that could function in the presence of CO and propene and, hence, could be used in a tandem reactor to enable the gas-phase conversion of propene and synthesis gas to butanol. To this end, we investigated the activity of silica-supported Shvo’s catalyst (Shvo/SiO2) for the gas-phase hydrogenation of butanal. Experiments were performed to determine the kinetics of n- and iso-butanal hydrogenation. The apparent activation energies and the apparent partial pressure dependencies of n- and iso-butanal, H2, and CO on the rates of n- and iso-butanol formation were determined. A mechanism for butanal hydrogenation was proposed to rationalize the observed kinetics and some of the reaction intermediates were observed by in situ infrared and 31P MAS NMR spectroscopy. It was found that Shvo/SiO2 was inhibited by SX (SX = sulfoxanthphos) and CO, and is inactive for alkene hydrogenation. The tandem catalytic conversion of propene and synthesis gas to butanol was then carried out using a SX-Rh supported ionic liquid phase (SILP) catalyst to promote the hydroformylation of propene to butanal and Shvo/SiO2 to promote the hydrogenation of butanal to butanol. The rate expressions describing the kinetics of each of the catalysts were then used to predict operating conditions required to achieve high conversion of propene to butanol. Under the most favorable conditions examined (H2/CO = 10), an overall yield of 13% to butanol was achieved with 15% propene conversion and 90% aldehyde conversion at a temperature of 413 K.Download high-res image (59KB)Download full-size image
Co-reporter:Zhenmeng Peng, Ferenc Somodi, Stig Helveg, Christian Kisielowski, Petra Specht, Alexis T. Bell
Journal of Catalysis (February 2012) Volume 286() pp:22-29
Publication Date(Web):1 February 2012
DOI:10.1016/j.jcat.2011.10.008
The formation of graphene layers on MgO-supported Pt nanoparticles was studied by both in situ and ex situ high-resolution transmission electron microscopy (HRTEM). The HRTEM images indicate that graphene sheets grow from steps in the surface of Pt nanoparticles. The subsequent morphology of the graphene sheets is a strong function of Pt particle size. For particles less than ∼6 nm in diameter, the graphene sheets form nanotubes or move from the surface of Pt particles and accumulate on the MgO support. Complete particle envelopment by multiple graphene layers was only observed for particle greater than ∼6 nm in diameter. The observed dependence of graphene morphology on Pt nanoparticle size and shape is associated with the strain energy generated between graphene layers during their growth and the overall free energy of the graphene-Pt system.Graphical abstractThe formation of graphene layers on MgO-supported Pt nanoparticles was studied by both in situ and ex situ high-resolution transmission electron microscopy (HRTEM), which indicate that graphene sheets grow from steps in the surface of Pt nanoparticles and the subsequent morphology of the graphene sheets is a strong function of Pt particle size.Download high-res image (94KB)Download full-size imageHighlights► In situ and ex situ combined HRTEM study on graphene formation on Pt/MgO model catalyst. ► Direct observation of graphene growth from Pt steps. ► Pt particle size-affected graphene morphology and in-depth discussion of the growth mechanism.
Co-reporter:Zheng Zhai, Andrew “Bean” Getsoian, Alexis T. Bell
Journal of Catalysis (December 2013) Volume 308() pp:25-36
Publication Date(Web):1 December 2013
DOI:10.1016/j.jcat.2013.05.008
•The activity Bi1−x/3V1−xMoxO4 for propene oxidation to acrolein exhibits a maximum at x = 0.45.•XANES measurements reveal that only Mo and V undergo reduction during reaction, but not Bi.•The observed pattern of activity with catalyst composition can be interpreted in terms of three types of active sites.•Rate parameters for each type of active site are determined and then used to describe the overall activity of Bi1−x/3V1−xMoxO4.We report the results of a systematic investigation of the kinetics of propene oxidation to acrolein over Bi1−x/3V1−xMoxO4. BET isotherms were measured to determine catalyst surface area, and powder X-ray diffraction was used to characterize the bulk structure. Further characterization by X-ray absorption near-edge spectroscopy (XANES) was used to determine the oxidation states of Bi, Mo, and V before and after exposure of the catalyst to propene at 713 K. We find that, contrary to previous discussions of the mechanism of propene oxidation on Bi1−x/3V1−xMoxO4, Bi remains in the 3+ state and only V and Mo undergo reduction and oxidation during reaction. The kinetics of propene oxidation were examined to establish the activation barrier for acrolein formation, and how the partial pressure dependences on propene and oxygen change with the value of x. The data obtained from this study were then used to propose a generalized model for the kinetics of propene oxidation over Bi1−x/3V1−xMoxO4 that is consistent with our findings about the reducibility of the three metallic elements in the oxide. According to this model, vanadium and molybdenum are randomly distributed to form three types of sites each associated with its own rate parameters. MoV sites are found to exhibit the highest activity. The proposed model provides a good description of the experimental data for all catalyst formulations examined, for a range of propene and oxygen partial pressures, and for temperatures above 653 K.Graphical abstractThe oxidation of propene to acrolein on Bi1−x/3V1−xMoxO4 was investigated in a systematic manner to identify the role of composition on catalyst activity and selectivity. The apparent rate coefficient kapp exhibits a maximum for 0 < x < 1. This pattern is very well described by a model involving three types of active sites.Download high-res image (86KB)Download full-size image
Co-reporter:Anton N. Mlinar, Guillaume B. Baur, Gerry G. Bong, Andrew “Bean” Getsoian, Alexis T. Bell
Journal of Catalysis (December 2012) Volume 296() pp:156-164
Publication Date(Web):1 December 2012
DOI:10.1016/j.jcat.2012.09.010
The oligomerization of propene was investigated over a series of nickel ion-exchanged Na-X zeolites with varying Ni loadings. Catalyst characterization by temperature-programmed reduction, elemental analysis, and XANES indicates that all of the exchanged Ni is present as Ni2+ that charge-compensates two exchange sites. The selectivity to propene oligomers remained greater than 98% for all Ni–Na-X catalysts with dimers being the main product. In contrast, the activity of Ni–Na-X was determined to depend strongly on Ni loading. At low to intermediate Ni loadings, the catalyst activates, reaches a maximum activity, and then deactivates with further time on stream. The rates of activation and deactivation are functions of the Ni content in the zeolite and both increase with increasing Ni loading. Stable activity was achieved for low Ni loadings (<0.6 wt%) by rapidly activating and deactivating the catalyst in propene at elevated temperature. The rate of propene dimerization measured under steady state conditions is first order in propene and characterized by an activation energy of 45 kJ mol−1. Activation of Ni–Na-X is attributed to migration of the Ni2+ cations from hexagonal prisms of the zeolite into the supercage where the cations form a catalytically active Ni2+–olefin complex. Deactivation is proposed to occur via the reaction of two nearby Ni–olefin complexes leading to the deactivation of both sites. A model for the dynamics of activation and deactivation and for the dimerization of propene to hexene is proposed. This model provides a satisfactory description of the effects of propene partial pressure and Ni loading on the rate of propene dimerization as a function of time on stream.Graphical abstractThe activity of Ni-exchanged Na-X zeolite for propene oligomerization increases as Ni2+ cations migrate from inside the hexagonal cages to the supercages where they are stabilized as Ni–propene complexes, but deactivation can occur when these species react together.Download high-res image (162KB)Download full-size imageHighlights► The activation of Ni–Na-X for propene oligomerization depends on Ni loading. ► Activation occurs via migration of Ni2+ cations from hexagonal prisms to zeolite supercages. ► Deactivation occurs via a two-site mechanism between nearby active sites. ► A model is proposed to account for catalyst activation, deactivation, and propene dimerization.
Co-reporter:David G. Hanna, Sankaranarayanapilla Shylesh, Sebastian Werner, Alexis T. Bell
Journal of Catalysis (August 2012) Volume 292() pp:166-172
Publication Date(Web):1 August 2012
DOI:10.1016/j.jcat.2012.05.011
An investigation of the kinetics of propene hydroformylation in the gas phase has been conducted over a silica-supported Rh–sulfoxantphos complex stabilized by the ionic liquid [bmim][OctSO4]. The reaction temperature was found to have a strong effect on the kinetics of n- and iso-butanal formation. For both products, it was observed that increasing the temperature decreased the apparent activation energy, altered the reaction orders with respect to reactants, and decreased the molar ratio of n- to iso-butanal. The observed changes in the kinetics are discussed in terms of the generally accepted mechanism for olefin hydroformylation and are attributed to a change in the rate-determining step (RDS). It is concluded that at low temperature, the RDS is alkene insertion into an Rh–H bond but becomes the oxidative addition of H2 at high temperature. The change in the RDS is rationalized in terms of a change in the elementary step with the largest Gibbs free energy of activation (ΔG‡). A greater loss in entropy for the oxidative addition of H2 over alkene insertion causes the ΔG‡ of the oxidative addition to be greater than the ΔG‡ of alkene insertion at high temperature.Graphical abstractAn investigation of the kinetics of propene hydroformylation in the gas phase has been conducted over a supported ionic liquid-phase (SILP) Rh–sulfoxantphos catalyst.Download high-res image (73KB)Download full-size imageHighlights► Kinetic orders with respect to H2 and CO are dependent upon reaction temperature. ► Reaction temperature is a tunable parameter to control the regioselectivity. ► The rate-determining step is alkene insertion and oxidative addition of H2 at low and high temperature, respectively. ► Derived rate expressions are consistent with kinetic studies and can be used to adequately model the experimental data.
Co-reporter:Arne Dinse, Max Aigner, Markus Ulbrich, Gregory R. Johnson, Alexis T. Bell
Journal of Catalysis (April 2012) Volume 288() pp:104-114
Publication Date(Web):1 April 2012
DOI:10.1016/j.jcat.2012.01.008
An investigation has been carried out of the effects of Mn promotion on the activity and product selectivity of Co/SiO2 for Fischer–Tropsch Synthesis (FTS). All experiments were conducted with an H2/CO feed ratio of 2.0 at either 1 atm or 10 atm and a temperature of 493 K. At 1 atm, the rate of CO consumption decreased with CO conversion, whereas the selectivities to all products were unaffected. However, the olefin to paraffin (O/P) ratio of the C2C4 fraction decreased with increasing CO conversion due to increased hydrogenation of olefins with increasing space time. Mn promotion increased the rate of CO consumption at Mn/Co ratios of 0.05, decreased the selectivity to methane, and increased the selectivity to C5+ products, but had no effect on the selectivity to C2C4 products. Raising the pressure to 10 atm increased the CO consumption rates for both unpromoted and Mn-promoted Co/SiO2, but the rate of reaction was lower for the Mn-promoted catalyst due to a higher level of CO inhibition. CO conversion at 10 atm had no effect on the rate of CO consumption for Co/SiO2 and caused a slight increase in the rate for Mn-promoted Co/SiO2. The intrinsic O/P ratio was higher at 10 atm than at 1 atm. In contrast to what was observed at 1 atm, at 10 atm, C2C4 of the C2C4 fraction incorporated into growing hydrocarbon chain, leading to a decrease in C1C4 selectivities and an increase in C5+ selectivities with increasing CO conversion. The observed effects of Mn promotion on catalyst activity and product selectivity are discussed in terms of the mechanisms for CO hydrogenation and hydrocarbon chain growth.Graphical abstractMn-promotion of Co/SiO2 affects the distribution of products by suppressing the formation methane and enhancing the formation of C5+ products as a consequence of olefin reincorporation.Download high-res image (58KB)Download full-size imageHighlights► Mn promotion of Co/SiO2 affects both its activity and selectivity for Fischer–Tropsch Synthesis (FTS). ► The effects of Mn are strongly dependent on Mn/Co ratio, reaction pressure, and CO conversion. ► At 10 atm, Mn promotion at Mn/Co = 0.5 suppresses methane selectivity and enhances C5+ selectivity.
Co-reporter:Anton N. Mlinar, Paul M. Zimmerman, Fuat E. Celik, Martin Head-Gordon, Alexis T. Bell
Journal of Catalysis (April 2012) Volume 288() pp:65-73
Publication Date(Web):1 April 2012
DOI:10.1016/j.jcat.2012.01.002
The oligomerization of propene was investigated over H-MFI zeolites with varying Si/Al ratios. For a constant space time per active site, the conversion of propene as well as the selectivity to products of different carbon number was affected by the density of the sites within the zeolite. In particular, as the Si/Al ratio decreased, corresponding to an increase in site proximity, the rate of oligomerization per site decreased but the selectivity to dimers relative to cracking products increased. These effects were shown to arise from the effects of molecular crowding on the rate coefficient for propene trimer formation and were confirmed by quantum chemical analysis of the energetics of propene oligomerization. It was found that the activation for propene dimerization is unaffected by the presence of oligomers on nearby sites, but the activation energy for propene trimerization relative to desorption of hexene increases by 19 kcal mol−1 when two next nearest neighbor sites are occupied by oligomers. In situ IR spectroscopy observations showed the buildup of aromatic species with time-on-stream. The accumulation of these species increases with decreasing Si/Al ratio, suggesting that increasing proximity of Brønsted-acid sites enhances the formation of aromatic species.Graphical abstractThe zeolite Si/Al ratio was observed to affect the activity and selectivity of propene oligomerization over H-MFI. Both a kinetic model as well as a quantum mechanic/molecular mechanic model illustrate that this effect is caused by the proximity of adsorbed oligomers restricting the formation of the trimer. This causes the rate coefficient for dimer desorption relative to the rate coefficient for trimer formation to increase with decreasing Si/Al ratio explaining the observed activity and selectivity behavior.Download high-res image (76KB)Download full-size imageHighlights► Decreasing MFI Si/Al ratio increases dimer selectivity but decreases propene conversion. ► Kinetic analysis indicates that change in selectivity and activity is caused by change in relative rate coefficients for trimerization and dimer desorption. ► QM/MM model indicates that steric crowding of nearby adsorbed dimer species increases activation energy of trimerization faster than dimer desorption. ► Larger amounts of aromatics/Al were produced in the more sterically crowded, low Si/Al ratio zeolites.
Co-reporter:Georges Siddiqi, Pingping Sun, Vladimir Galvita, Alexis T. Bell
Journal of Catalysis (9 September 2010) Volume 274(Issue 2) pp:200-206
Publication Date(Web):9 September 2010
DOI:10.1016/j.jcat.2010.06.016
The dehydrogenation of ethane and propane using a Pt catalyst supported on a novel Mg(Ga)(Al)O mixed oxide support was investigated. Catalyst performance is strongly dependent on Ga content in the support, a peak in activity for both ethane and propane dehydrogenation occurs at Ga/Pt = 1.4–5.4, and selectivity is a monotonic function of Ga/Pt, reaching nearly 100% at Ga/Pt = 5.4. The addition of hydrogen to the feed resulted in a peak in activity with respect to H2/alkane. The increase in dehydrogenation rate with H2 addition is attributed to H-atom-assisted dehydrogenation of alkyl species formed upon dissociative adsorption of the reactant alkane. Beyond the peak in activity with H2 addition, a further increase in H2 feed concentration contribute to alkene hydrogenation, thereby reducing the net rate of dehydrogenation. Hydrogen addition to the feed, however, had relatively little effect on alkene selectivity, which remained near 100%. The presence of Ga also suppressed coke formation. Interestingly, less coke was formed during propane dehydrogenation than ethane dehydrogenation, and no correlation was found between coke formation and catalyst deactivation. Thus, the extent of deactivation was lower for ethane than propane dehydrogenation, whereas the amount of coke deposited was higher in the former case. Since the amount of carbon deposited as coke is higher than the amount of exposed Pt, it is concluded that most of the coke resides on the support, and that only a small amount resides on the Pt particles. The higher level of deactivation seen during propane versus ethane dehydrogenation is attributed to a higher coverage of Pt by coke precursors derived from propane than ethane.Mg(Ga)(Al)O-supported Pt reduced at 873 K exhibits high activity, selectivity, and stability for the dehydrogenation of ethane and propane. Best performance is achieved for bulk Ga/Pt ratio of 5.4. The presence of Ga significantly reduces the deposition of coke.Download high-res image (73KB)Download full-size image
Co-reporter:Pingping Sun, Georges Siddiqi, Miaofang Chi, Alexis T. Bell
Journal of Catalysis (9 September 2010) Volume 274(Issue 2) pp:192-199
Publication Date(Web):9 September 2010
DOI:10.1016/j.jcat.2010.06.017
A novel approach is described for preparing Ga-promoted Pt particles for the dehydrogenation of light alkanes to alkenes. The modifying element, Ga, was introduced by transference from the support, a calcined Mg(Ga)(Al)O hydrotalcite-like material. Pt nanoparticles were dispersed onto the calcined Mg(Ga)(Al)O starting from an organometallic precursor, followed by reduction. The formation of PtGa alloy particles is dependent on reduction temperature. Reduction at 723 K produces mainly metallic Pt particles. The average diameter of the Pt nanoparticles increased from 1.4 nm to 2.2 nm with increasing Ga content, and decreasing Al content of the support, demonstrating the importance of support Al atoms in stabilizing the dispersion of Pt. After reduction at 773–873 K, PtGa alloys were observed. It is proposed that at high reduction temperatures, H atoms formed on the surface of the metal particles spill over onto the support where they reduce Ga3+ cation to atomic Ga, which then interacts with the supported Pt to form PtGa alloys. The activity, selectivity and stability of Pt/Mg(Ga)(Al)O catalysts for ethane and propane dehydrogenation are described in the second part of this study (G. Siddiqi, P. Sun, V. Galvita, A.T. Bell, Journal of catalysis (2010), doi:10.1016/j.jcat.2010.06.016 [40]).PtGa bimetallic catalysts have been prepared by depositing Pt nanoparticles on the surface hydrotalcite-like supports, Mg(Ga)(Al)O. Upon reduction at 873 K, Ga3+ cations at the support surface are reduced to Ga, which then interacts with the Pt nanoparticles to form PtGa alloys. Evidence for PtGa is obtained from EXAFS and STEM-EDX.Download high-res image (56KB)Download full-size image
Co-reporter:Fuat E. Celik, Taejin Kim, Anton N. Mlinar, Alexis T. Bell
Journal of Catalysis (9 September 2010) Volume 274(Issue 2) pp:150-162
Publication Date(Web):9 September 2010
DOI:10.1016/j.jcat.2010.06.015
In situ IR spectroscopy was used to observe the intermediates formed on zeolites FAU and MFI during the synthesis of methyl methoxyacetate (MMAc) via carbonylation of dimethoxymethane (DMM) and the disproportionation of DMM to dimethyl ether (DME) and methyl formate (MF). Both reactions are initiated by the reaction of DMM with the Brønsted acid protons of the zeolite to form methanol and methoxymethoxy groups (MMZ). The latter species then undergoes one of two processes – carbonylation to form methoxyacetyl species, the precursors to MMAc, or reaction with DMM, resulting in DMM disproportionation. Surface intermediates for both DMM carbonylation and disproportionation respond to changes in reaction conditions in a manner consistent with observed steady-state kinetics. DMM carbonylation occurred more rapidly in the presence than absence of physisorbed DMM, a phenomenon attributed to solvation of the carbocationic transition state involved in the addition of CO to MMZ predicted by DFT calculations. The surface concentration of the methoxyacetyl species at steady state was 10 times smaller on FAU than on MFI, consistent with the higher rate of DMM carbonylation on FAU. Rate expressions for the formation of each product, based on the proposed mechanisms, in combination with a suitable set of rate coefficients, give a good description of the experimentally observed dependences of the rates of product formation on temperature and the feed partial pressures of CO and DMM.The mechanisms of dimethoxymethane (DMM) carbonylation and disproportionation over zeolites FAU and MFI were investigated using in situ IR spectroscopy. Rate expressions based upon the reaction pathways deduced from these studies describe the rates of product formation as functions of the reaction temperature and the feed partial pressures of CO and DMM. For both FAU and MFI, the rate of methoxyacetate (MMAc) formation, the product of DMM carbonylation, increased with the intensity of the IR peak for adsorbed methoxymethyl acyl species, the precursor to MMAc, consistent with the proposed mechanism of DMM carbonylation.Download high-res image (110KB)Download full-size image
Co-reporter:Vladimir Galvita, Georges Siddiqi, Pingping Sun, Alexis T. Bell
Journal of Catalysis (4 May 2010) Volume 271(Issue 2) pp:209-219
Publication Date(Web):4 May 2010
DOI:10.1016/j.jcat.2010.01.016
The dehydrogenation of ethane to ethene on Sn-promoted Pt supported on calcined hydrotalcite, PtSn/Mg(Al)O, was investigated with the aim of understanding the effects of Sn on the local environment of the dispersed Pt, the catalyst activity and selectivity for dehydrogenation, and the formation of coke. The origins of methane, the primary byproduct of ethane dehydrogenation were also investigated as a part of this study. Pt/Mg(Al)O was reacted with tetra-n-butyl tin in order to introduce Sn selectively to the dispersed Pt particles. High-resolution TEM revealed the formation of a PtSn bimetallic phase upon introduction of Sn. The activity of PtSn/Mg(Al)O for ethane dehydrogenation at 873 K was highest for a Sn/Pt ratio of 0.3, whereas the ethene selectivity increased monotonically with increasing Sn/Pt ratio, reaching 100% for Sn/Pt = 0.4. Sn promotion also significantly decreased the deposition of coke. Addition of H2 to the feed enhanced the formation of ethene for H2/C2H6 ratios up to 0.58 for Pt/Mg(Al)O and 0.25 for PtSn/Mg(Al)O, but for higher ratios the product concentration of ethene decreased and approached that determined thermodynamically. The ethene selectivity decreased dramatically with increasing H2/C2H6 ratio for Pt/Mg(Al)O but only slightly for PtSn/Mg(Al)O. Coke formation was suppressed considerably by H2 addition to the feed, particularly for PtSn/Mg(Al)O. Isotopic tracer studies revealed that methane formation resulted primarily from the readsorption of ethene. A mechanism is proposed for ethane dehydrogenation and is used to interpret the effects of Sn promotion and the addition of H2 to the feed.The effects of Sn on the dehydrogenation of ethane on Pt supported on calcined hydrotalcite (Pt/Mg(Al)O) were investigated. Sn was found to form an alloy with Pt and to enhance ethene selectivity and decrease coke deposition. Adsorption of ethene was identified as the primary source of CHx (x = 1–3) species that are precursors to the formation of methane and coke.Download high-res image (62KB)Download full-size image
Co-reporter:Joseph Zakzeski, Irene S. Fan, Alexis T. Bell
Applied Catalysis A: General (31 May 2009) Volume 360(Issue 1) pp:33-37
Publication Date(Web):31 May 2009
DOI:10.1016/j.apcata.2009.02.042
Co-reporter:Yihua Zhang, Daniel N. Briggs, Emiel de Smit, Alexis T. Bell
Journal of Catalysis (25 October 2007) Volume 251(Issue 2) pp:443-452
Publication Date(Web):25 October 2007
DOI:10.1016/j.jcat.2007.07.018
The aim of this work was to establish the effects of zeolite structure/chemical composition on the activity and selectivity of Cu-exchanged Y (Si/Al = 2.5), ZSM-5 (Si/Al = 12), and Mordenite (Si/Al = 10) for the oxidative carbonylation of methanol to DMC. Catalysts were prepared by solid-state ion-exchange of the H-form of each zeolite with CuCl and were then characterized by FTIR and X-ray absorption spectroscopy (XAS). The XANES portion of the XAS data showed that all of the copper was present as Cu+ cations, and analysis of the EXAFS portion of the data shows the Cu+ cations had a CuO coordination number of ∼2.1 on Cu-Y and ∼2.7 on Cu-ZSM-5 and Cu-MOR. Dimethyl carbonate (DMC) was observed as the primary product when a mixture of CH3OH/CO/O2 was passed over Cu-Y, whereas dimethoxy methane was the primary product over Cu-ZSM-5 and Cu-MOR. The higher activity and selectivity of Cu-Y for the oxidative carbonylation of methanol can be attributed to the weaker adsorption of CO on the Cu+ cations exchanged into Y zeolite. In situ IR observations revealed that under reaction conditions, adsorbed CO was displaced by methoxide groups bound to the Cu+ cations. The kinetics of DMC synthesis suggests that the rate-limiting step in the formation of this product was the insertion of CO into CuOCH3 bonds. The yield of DMC decreased with methanol conversion, likely due to the hydrolysis of DMC to methanol and carbon dioxide.
Co-reporter:Yihua Zhang, Alexis T. Bell
Journal of Catalysis (25 April 2008) Volume 255(Issue 2) pp:153-161
Publication Date(Web):25 April 2008
DOI:10.1016/j.jcat.2008.01.033
The mechanism of dimethyl carbonate (DMC) synthesis from oxidative carbonylation of methanol over Cu-exchanged Y zeolite has been investigated using in situ infrared spectroscopy and mass spectrometry under transient-response conditions. The formation of DMC is initiated by reaction of molecularly adsorbed methanol with oxygen to form either mono- or di-methoxide species bound to Cu+ cations. Reaction of the mono-methoxide species with CO produces monomethyl carbonate (MMC) species. DMC is formed via two distinct reaction pathways—CO addition to di-methoxide species or by reaction of methanol with MMC. The rate-limiting step in DMC synthesis is found to be the reaction of CO with mono-methoxide or di-methoxide species. The first of these reactions produces MMC, which then reacts rapidly with methanol to produce DMC, whereas the second of these reactions produces DMC directly. Formaldehyde was identified as an intermediate in the formation of dimethoxy methane (DMM) and methyl formate (MF). Both byproducts are thought to form via a hemiacetal intermediate produced by the reaction of methanol with adsorbed formaldehyde at a Cu+ site.
Co-reporter:Fuat E. Celik, Tae-Jin Kim, Alexis T. Bell
Journal of Catalysis (22 March 2010) Volume 270(Issue 1) pp:185-195
Publication Date(Web):22 March 2010
DOI:10.1016/j.jcat.2009.12.021
This work reports on the effects of zeolite framework type and Si/Al ratio on the carbonylation of dimethoxymethane (DMM) to produce methyl methoxyacetate (MMAc). Faujasite (FAU), ZSM-5 (MFI), Mordenite (MOR) and Beta (BEA) showed very similar activity for DMM carbonylation. However, FAU had a very high selectivity to MMAc compared to MFI, MOR and BEA because of very low rates of dimethyl ether (DME) and methyl formate (MF) formation, by-products of the disproportionation of DMM. The high rate of DMM disproportionation observed for MFI, MOR and BEA is ascribed to the small pores of these zeolites, which facilitate a critical initial step in the formation of DME and MF. FER showed very low activity for both carbonylation and disproportionation. Increasing the Si/Al ratio for both FAU and MFI led to an increase in the turnover frequency for DMM carbonylation. It is proposed that the low rate of MMAc formation found at low Si/Al ratios is due to repulsive interactions occurring between adsorbed species located within the same supercage (FAU) or channel intersection (MFI).The role of zeolite structure and composition in the carbonylation of dimethoxymethane to methyl methoxyacetate was investigated at steady state. Faujasite (FAU) showed higher selectivity than medium-pore zeolites such as ZSM-5 (MFI). Carbonylation rates increased with increasing Si/Al ratio until one or fewer Al atoms were located in the supercage of FAU or the channel intersections of MFI.Download high-res image (71KB)Download full-size image
Co-reporter:Daniel N. Briggs, Kenneth H. Lawrence, Alexis T. Bell
Applied Catalysis A: General (15 September 2009) Volume 366(Issue 1) pp:
Publication Date(Web):15 September 2009
DOI:10.1016/j.apcata.2009.06.032
The synthesis of diethyl carbonate (DEC) by the oxidative carbonylation of ethanol was investigated using catalysts prepared by the dispersion of CuCl2 and PdCl2 on amorphous carbon promoted with KCl and NaOH. Catalysts were characterized extensively by XRD, XAFS, SEM and TEM with the aim of establishing their composition and structure after preparation, pretreatment, and use. It was observed that after preparation and pretreatment in He at 423 K copper is present almost exclusively as Cu(I), most likely in the form of [CuCl2]− anions, whereas palladium is present as large PdCl2 particles. Catalysts prepared exclusively with copper or palladium chloride are inactive for DEC synthesis, indicating that both components must be present together. Evidence from XANES and EXAFS suggests that the DEC synthesis may occur on [PdCl2−x][CuCl2]x species deposited on the surface of the PdCl2 particles. As-prepared catalysts exhibit an increase in DEC synthesis activity and selectivity with time on stream, but then reach a maximum activity and selectivity, followed by a slow decrease in DEC activity. The loss of DEC activity is accompanied by a loss in Cl from the catalyst and the appearance of paratacamite.The synthesis of diethyl carbonate by the oxidative carbonylation of ethanol was investigated using catalysts prepared by dispersing CuCl2 and PdCl2 on amorphous carbon. Evidence from XRD, XAFS, SEM and TEM indicated that copper is present as [CuCl2]− anions, whereas palladium exists as large PdCl2 particles. Together with catalysis data, these results suggest that DEC synthesis may occur on [PdCl2−x][CuCl2]x species deposited on the surface of PdCl2 particles.Download full-size image
Co-reporter:Daniel N. Briggs, Gerry Bong, Eric Leong, Kevin Oei, Gabriella Lestari, Alexis T. Bell
Journal of Catalysis (15 December 2010) Volume 276(Issue 2) pp:215-228
Publication Date(Web):15 December 2010
DOI:10.1016/j.jcat.2010.08.004
The oxidative carbonylation of ethanol to diethyl carbonate (DEC) has been investigated on catalysts prepared by dispersing CuCl2 and PdCl2 on activated carbon and carbon nanofibers. The objectives of this work were to establish the effects of support structure and pretreatment on the dispersion of the catalytically active components and, in turn, on the activity and selectivity of the catalyst for DEC synthesis. At the same surface loading of CuCl2 and PdCl2, partially oxidized carbon nanofibers resulted in a higher dispersion of the active components and a higher DEC activity than could be achieved on activated carbon. Catalyst characterization revealed that nearly atomic dispersion of CuCl2 and PdCl2 could be achieved on the edges of the graphene sheets comprising the carbon nanofibers. Over oxidation of the edges or their removal by heat treatment of the nanofibers resulted in a loss of catalyst activity. The loss of catalyst activity with time on stream could be overcome by the addition of ppm levels of CCl4 to the feed. While catalysts prepared with CuCl2 alone were active, a fivefold increase in activity was realized by using a PdCl2/CuCl2 ratio of 1/20. It is proposed that the Pd2+ cations interact with [CuCl2]− anions to form Pd[CuCl2]2 complexes that are stabilized through dative bonds formed with oxygen groups present at the edges of the graphene sheets of the support. A mechanism for DEC synthesis is discussed, and a role for the Pd2+ cations as part of this mechanism is proposed.PdCunClx species dispersed on carbon supports catalyze the oxidative carbonylation of ethanol to diethyl carbonate (DEC). Catalyst activity and selectivity are improved by oxidation of the carbon support before preparation, and catalyst stability can be achieved by the addition of ppm levels of CCl4 into the feed. It is proposed that the highest activity is exhibited by Cl-bridged [CuCl2]Pd[CuCl2] species.Download high-res image (73KB)Download full-size image
Co-reporter:N. Hansen, R. Krishna, J.M. van Baten, A.T. Bell, F.J. Keil
Chemical Engineering Science (15 April 2010) Volume 65(Issue 8) pp:2472-2480
Publication Date(Web):15 April 2010
DOI:10.1016/j.ces.2009.12.028
Rate expressions are vital for analysis, design and operation of chemical reactors. However, due to a simplified picture of the underlying physical processes, empirical power-law (PL) fits, and Langmuir–Hinshelwood (LH) expressions may not be reliable when extrapolated to conditions which were not included in the parameterization process. In the present work the extrapolation ability of LH and PL models are evaluated for the zeolite catalyzed alkylation of benzene with ethene. For this purpose, extrapolated data are compared to results obtained from a detailed continuum model based on a multiscale approach (Hansen et al., J. Phys. Chem. C, 113 (2009) 235–246). It is demonstrated that extrapolation is in particular questionable if the gas phase composition is outside the fitting range. A second purpose of the present work is the extension of our continuum model to include the dehydrogenation of ethane. The parameters describing adsorption and diffusion are obtained from Monte Carlo and molecular dynamics simulations, respectively. Reaction rate constants are derived from quantum chemical calculations and transition state theory. We have used the extended continuum model in the design equation of a fixed bed reactor and simulated the dehydroalkylation activity for different input conditions. Furthermore, the benefit from removing hydrogen from the reaction mixture using a membrane reactor is discussed.
Co-reporter:Niels Hansen, Andreas Heyden, Alexis T. Bell, Frerich J. Keil
Journal of Catalysis (10 June 2007) Volume 248(Issue 2) pp:213-225
Publication Date(Web):10 June 2007
DOI:10.1016/j.jcat.2007.03.015
The decomposition of N2O on dinuclear oxygen-bridged iron sites in Fe-ZSM-5 was simulated under steady-state conditions considering the reaction mechanism and the rate parameters proposed by Hansen et al. [J. Phys. Chem. C 111 (2007) 2092] on the basis of DFT calculations. The presence of low concentrations of water vapor in the feed stream (ppb to ppm levels) affects the calculated values for the apparent activation energy and the pre-exponential factor and thus can explain the wide variation in experimental values for these quantities, as well as the appearance of an apparent compensation effect. The activity of the dinuclear oxygen-bridged site was compared with that of the mononuclear iron site proposed earlier by Heyden et al. [J. Phys. Chem. B 109 (2005) 1857]; the latter was found to be slightly more active. Microkinetic models for both mononuclear and dinuclear iron sites were used to reproduce temperature-programmed reaction experiments reported for Fe-ZSM-5 samples with low and high iron content. This analysis leads to the conclusion that at very low Fe/Al ratios, mononuclear iron sites prevail, whereas at higher Fe/Al ratios, both mononuclear and dinuclear iron sites are likely to be present simultaneously.
Co-reporter:Shaji Chempath, Alexis T. Bell
Journal of Catalysis (1 April 2007) Volume 247(Issue 1) pp:119-126
Publication Date(Web):1 April 2007
DOI:10.1016/j.jcat.2007.01.012
A theoretical analysis was carried out of the mechanism and kinetics of methane oxidation to formaldehyde occurring on isolated molybdate species supported on silica. Both mono-oxo and di-oxo molybdate structures were used to represent the active centers. The energetics for each elementary reaction was determined from density functional theory calculations, and the entropy changes were determined from calculations based on statistical mechanics. The results of this analysis show that the mechanism based on di-oxo molybdate species agrees more closely with observed rates of methane oxidation than that based on mono-oxo molybdate species. It is also found that the formation of formaldehyde occurs via the reaction of methane with peroxide species formed via the adsorption of O2 on reduced MoIV centers. The extent of MoVI reduction to MoIV is well under 1% under reaction conditions, in good agreement with experimental observations.
Co-reporter:Zhenmeng Peng, Christian Kisielowski and Alexis T. Bell
Chemical Communications 2012 - vol. 48(Issue 13) pp:NaN1856-1856
Publication Date(Web):2011/12/14
DOI:10.1039/C2CC16962B
A novel method has been developed for preparing supported cubic platinum nanoparticles. Carbon monoxide and hydrogen are used to reduce platinum precursors present at a solid–gas interface and to control the shape of the growing Pt nanoparticles. By avoiding the use of any organic agents in the synthesis, cubic Pt particles free of hydrocarbons are formed, thereby avoiding possible contamination of the catalyst surface. The approach used is simple and readily scalable.
Co-reporter:Joseph Zakzeski, Sarah Burton, Andrew Behn, Martin Head-Gordon and Alexis T. Bell
Physical Chemistry Chemical Physics 2009 - vol. 11(Issue 42) pp:NaN9911-9911
Publication Date(Web):2009/08/21
DOI:10.1039/B906883J
A spectroscopic investigation of complexes used to catalyze the oxidative carbonylation of toluene to p-toluic acid was conducted. Rhodium complexes were analyzed by 103Rh and 13C NMR, UV-visible spectroscopy, and infrared spectroscopy. In the presence of vanadium and oxygen, the resting state of the Rh-catalyst was found to exist as a Rh(III) complex with carbonyl and trifluoroacetate ligands, consistent with the structure Rh(CO)2(TFA)3. The 13C NMR spectrum of Rh(13CO)2(TFA)3 complex exhibited a carbonyl peak with an unusual degree of shielding, which resulted in the appearance of the carbonyl peak at an unprecedented upfield position in the 13C NMR spectrum. This shielding was caused by interaction of the carbonyl group with the trifluoroacetate ligand. In the absence of oxygen, the Rh(III) complex reduced to Rh(I), and the reduced form exhibited properties resembling the catalyst precursor. Structures and spectroscopic properties calculated using density functional theory agreed closely with the experimental results. The vanadium co-catalyst used to reoxidize Rh(I) to Rh(III) was similarly characterized by 51V NMR and UV-visible spectroscopy. The oxidized species corresponded to [(VO2)(TFA)]2, whereas the reduced species corresponded to (VO)(TFA)2. The spectroscopic results obtained in this study confirm the identity of the species that have been proposed to be involved in the Rh-catalyzed oxidative carbonylation of toluene to toluic acid.
Co-reporter:Arne Dinse, Reinhard Schomäcker and Alexis T. Bell
Physical Chemistry Chemical Physics 2009 - vol. 11(Issue 29) pp:NaN6124-6124
Publication Date(Web):2009/05/11
DOI:10.1039/B821131K
The oxidative dehydrogenation (ODH) of ethane on alumina-supported vanadia was investigated with the aim of understanding the effects of lattice oxygen and vanadium oxidation state on the catalyst ODH activity and ethene selectivity. Transient-response experiments were carried out with both a fully oxidized sample of 10 wt% VOx/Al2O3 (7 V nm−2) and a sample that had been partially reduced in H2. The experimental results were analyzed to determine the rate coefficients for ethane ODH, k1, and ethene combustion, k3. The rate of ODH was found to depend solely on the concentration of reactive oxygen in the catalyst, but not on the means by which this oxygen concentration was attained (i.e., by H2versus C2H6 reduction). On the other hand, the ethene selectivity observed at a given concentration of active oxygen was found to depend on the composition of the reducing agent, higher ethene selectivities being observed when H2, rather than C2H6, was used as the reducing agent. It is proposed that the higher ethene selectivity achieved by H2versus C2H6 reduction might be due to a lower ratio of V4+ to V3+ cations attained upon reduction in H2 for a given extent of V5+ reduction. This interpretation is based on the hypothesis that ethene combustion is initiated by C2H4 adsorption on Vn+ cations present at the catalyst surface and that the strength of adsorption decreases in the order V5+ > V4+ > V3+ consistent with the decreasing Lewis acidity of the cations.
Co-reporter:Kristopher R. Enslow and Alexis T. Bell
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 5) pp:NaN2847-2847
Publication Date(Web):2015/03/16
DOI:10.1039/C5CY00077G
A number of Lewis acid catalysts were screened for their effectiveness in converting both xylose and glucose in aqueous media to furfural and 5-HMF, respectively. While other catalysts were found to be more active, SnCl4 was identified as the most selective Lewis acid. Hydrolysis of SnCl4 was observed at various concentrations and temperatures resulting in the production of Brønsted acidic protons in a 3.5:1 ratio to Sn4+ at all SnCl4 concentrations above 60 °C. As a consequence, there was no need to add a Brønsted acid in order to promote the dehydration of either xylose or glucose. SnCl4-promoted isomerization/dehydration of xylose and glucose at 140 °C in water resulted in conversions of 55% and 33%, respectively, after 2 h of reaction, and furfural and 5-HMF selectivities of up to 58% and 27%, respectively. Significant conversion of sugars to humins was observed in both cases, and in the case of glucose, degradation of 5-HMF to levulinic and formic acids was also noted. The effects of secondary reactions could be greatly suppressed by extraction of the furanic product as it was produced. Using n-butanol as the extracting agent, xylose and glucose conversions of 90% and 75%, respectively, were observed after 5 h of reaction, and the selectivities to furfural and 5-HMF increased to 85% and 69%, respectively. Small additional increases in the furfural and 5-HMF selectivities were obtained by adding LiCl to the aqueous phase without much effect on the conversion of either sugar. In this case, the selectivities to furfural and 5-HMF were 88% and 72%, respectively, after 5 h of reaction at 140 °C.
Co-reporter:Evan M. W. Rumberger, Hyun S. Ahn, Alexis T. Bell and T. Don Tilley
Dalton Transactions 2013 - vol. 42(Issue 34) pp:NaN12247-12247
Publication Date(Web):2013/07/12
DOI:10.1039/C3DT51472B
Adsorption of a dinuclear μ-oxo bridged Mn complex onto mesoporous silica was observed when SBA15 was treated with an acetonitrile solution of [Mn2(μ-O)2Cl(μ-O2CCH3)(H2O)(bpy)2](NO3)2 (1). This complex was immobilized via the displacement of NO3− into solution, and characterization by spectroscopic (DRIFTS and DRUV-vis) and magnetic data indicates that the intact dication is electrostatically bound to the silica surface. Loadings of up to 4.1% by weight of [Mn2(μ-O)2Cl(μ-O2CCH3)(H2O)(bpy)2]2+ were achieved. TEM images of the grafted material revealed retention of the mesoporous structure of SBA15, and no clusters of manganese greater than ca. 10 nm were observed. The SBA15-supported dimanganese complex functions as a catalyst for the oxidation of H2O with (NH4)2Ce(NO3)6 as stoichiometric oxidant. In contrast, homogenous aqueous solutions of 1 do not evolve oxygen upon treatment with (NH4)2Ce(NO3)6. Labeling studies with H218O confirm that the oxygen formed in this catalysis is derived from water. Monitoring the O2 evolution allowed determination of an initial rate for the catalysis (TOFi = 1.1 × 10−3 s−1). These studies also reveal a first order dependence on manganese surface concentration, and a zero order rate dependence for (NH4)Ce(NO3)6. Spectroscopic investigations were employed to investigate the difference in activities between dissolved and supported dimanganese complexes.
.jpg)

















![4-Pyridinamine, N-methyl-N-[3-(triethoxysilyl)propyl]-](http://img.cochemist.com/ccimg/128900/128823-45-0.png)
![4-Pyridinamine, N-methyl-N-[3-(triethoxysilyl)propyl]-](http://img.cochemist.com/ccimg/128900/128823-45-0_b.png)
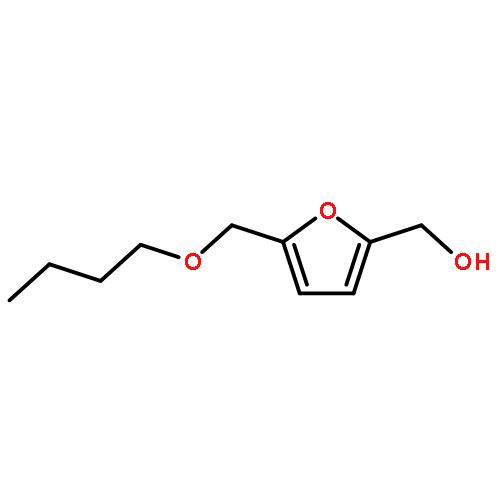




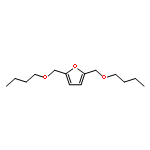
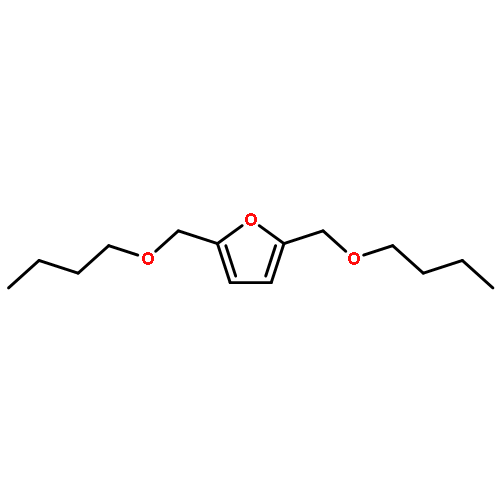




![1-Propanesulfonic acid, 3-[[3-(trihydroxysilyl)propyl]thio]-](http://img.cochemist.com/ccimg/88700/88683-98-1.png)
![1-Propanesulfonic acid, 3-[[3-(trihydroxysilyl)propyl]thio]-](http://img.cochemist.com/ccimg/88700/88683-98-1_b.png)


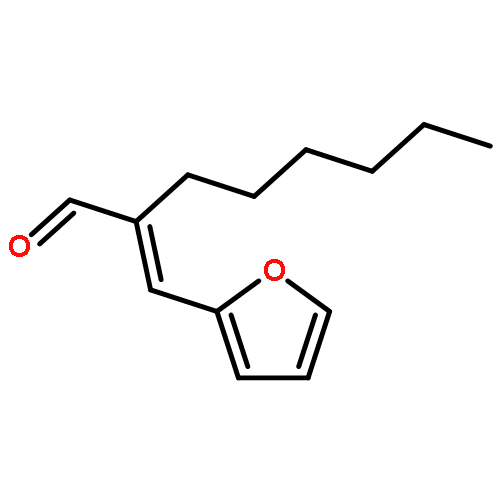




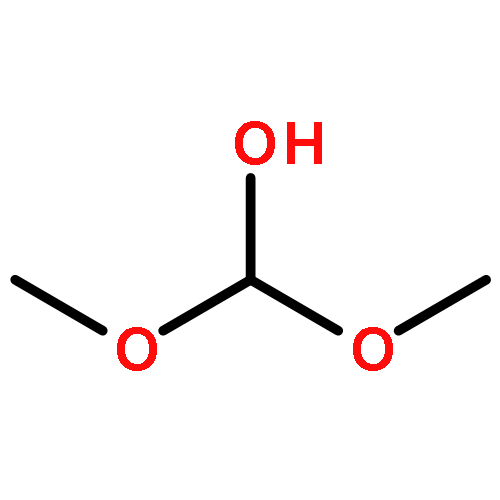

















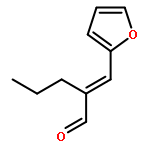
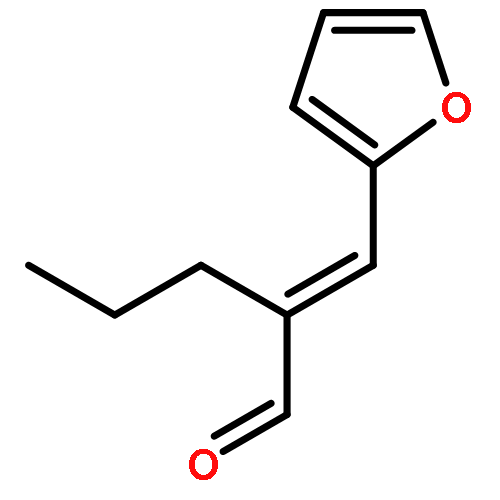

































![Bicyclo[1.1.0]butane](http://img.cochemist.com/ccimg/200/157-33-5.png)
![Bicyclo[1.1.0]butane](http://img.cochemist.com/ccimg/200/157-33-5_b.png)