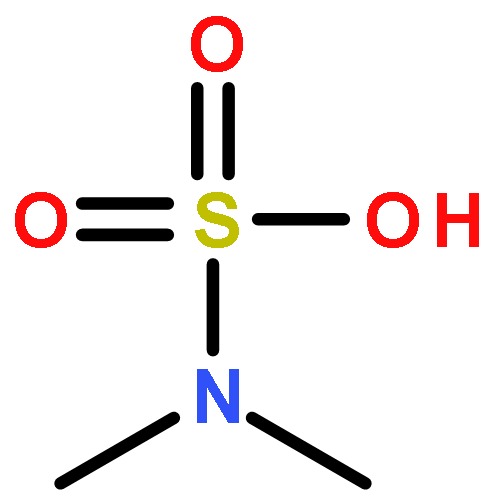
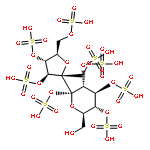
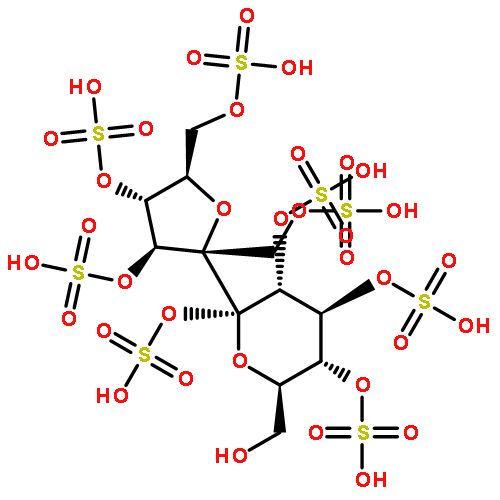
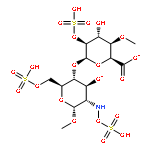
Co-reporter:Daniel K. Afosah, Rami A. Al-Horani, Nehru Viji SankaranarayananUmesh R. Desai
Journal of Medicinal Chemistry 2017 Volume 60(Issue 2) pp:
Publication Date(Web):December 15, 2016
DOI:10.1021/acs.jmedchem.6b01474
Although plasmin inhibitors could be used in multiple disorders, their use has been restricted to preventing blood loss in hemostatic dysregulation because of poor efficacy and adverse effects of current agents. We reasoned that a new class of direct inhibitors that offer better efficacy, selectivity, and safety could be discovered by exploiting allosterism in plasmin, a protease homologous to other allosteric serine proteases. We report on the synthesis, biological activity, and mechanism of action of a group of small molecules, called non-saccharide glycosaminoglycan mimetics (NSGMs), as direct allosteric plasmin inhibitors. Our results show that distinct NSGMs selectively inhibit human full-length plasmin. The molecule inhibited clot lysis, alluding to its promise as an allosteric regulator of plasmin. We show that direct allosteric inhibition of plasmin could led to new antifibrinolytic agent(s) that may exhibit better efficacy, potency, selectivity, and safety in comparison to current therapy.
Co-reporter:Tien M. Truong, Hua Li, Sneha Dhapare, Umesh R. Desai, Nobert F. Voelkel, Masahiro Sakagami
Pulmonary Pharmacology & Therapeutics 2017 Volume 45(Volume 45) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.pupt.2017.06.007
Induced lung cell death and impaired hypoxia-inducible factor-1α (HIF-1α) and vascular endothelial growth factor (VEGF) signaling are proposed as a pathobiologic mechanism for alveolar structural destruction and loss in emphysema. We hypothesized that our sulfated dehydropolymer of caffeic acid, CDSO3, exerts anti-cell death activities and therapeutic interventions in emphysema by virtue of Fe2+ chelation-based HIF-1α/VEGF stabilization and elevation. The Fe2+ chelating activity was determined in the chromogenic ferrozine-Fe2+ chelation inhibitory assay. The in vitro anti-cell death activities and their Fe2+ and HIF-1α dependence were assessed against a range of emphysematous insults in the lung endothelial (HMVEC-L) and epithelial (A549) cells. CDSO3 was spray-dosed to the lung for three weeks (day 1–21) in an in vivo rat model of apoptotic emphysema induced with a VEGF receptor antagonist SU5416. Post-treatment treadmill exercise endurance, airspace enlargement, and several lung biomarkers/proteins were measured. CDSO3 was a potent Fe2+ chelating molecule. At 10 μM, CDSO3 inhibited HMVEC-L and A549 cell death induced by histone deacetylase inhibition with trichostatin A, VEGF receptor blockade with SU5416, and cigarette smoke extract by 65–99%, which were all significantly opposed by addition of excess Fe2+ or HIF-1α inhibitors. As a potent elastase inhibitor and antioxidant, CDSO3 also inhibited elastase- and H2O2-induced cell death by 92 and 95%, respectively. In the rat model of SU5416-induced apoptotic emphysema, CDSO3 treatment at 60 μg/kg 1) produced 61–77% interventions against exercise endurance impairment, airspace enlargement [mean linear intercept] and oxidative lung damage [malondialdehyde activity]; 2) normalized the apoptotic marker [cleaved caspase-3]; 3) stimulated the VEGF signaling [VEGF receptor 2 phosphorylation] by 1.4-fold; and 4) elevated the HIF-1α and VEGF expression by 1.8- and 1.5-fold, respectively. All of these were consistent with CDSO3's Fe2+ chelation-based HIF-1α/VEGF stabilization and elevation against their pathobiologic deficiency, inhibiting lung cell death and development of apoptotic emphysema.
Co-reporter:Rio S. Boothello, Aurijit Sarkar, Vy My Tran, Thao Kim Nu Nguyen, Nehru Viji Sankaranarayanan, Akul Y. Mehta, AlHumaidi Alabbas, Spencer Brown, Alessandro Rossi, April C. Joice, Caitlin P. Mencio, Maritza V. Quintero, Balagurunathan Kuberan, and Umesh R. Desai
ACS Chemical Biology 2015 Volume 10(Issue 6) pp:1485
Publication Date(Web):March 5, 2015
DOI:10.1021/acschembio.5b00071
The structural diversity of natural sulfated glycosaminoglycans (GAGs) presents major promise for discovery of chemical biology tools or therapeutic agents. Yet, few GAGs have been identified so far to exhibit this promise. We reasoned that a simple approach to identify such GAGs is to explore sequences containing rare residues, for example, 2-O-sulfonated glucuronic acid (GlcAp2S). Genetic algorithm-based computational docking and filtering suggested that GlcAp2S containing heparan sulfate (HS) may exhibit highly selective recognition of antithrombin, a key plasma clot regulator. HS containing only GlcAp2S and 2-N-sulfonated glucosamine residues, labeled as HS2S2S, was chemoenzymatically synthesized in just two steps and was found to preferentially bind antithrombin over heparin cofactor II, a closely related serpin. Likewise, HS2S2S directly inhibited thrombin but not factor Xa, a closely related protease. The results show that a HS containing rare GlcAp2S residues exhibits the unusual property of selective antithrombin activation and direct thrombin inhibition. More importantly, HS2S2S is also the first molecule to activate antithrombin nearly as well as the heparin pentasaccharide although being completely devoid of the critical 3-O-sulfonate group. Thus, this work shows that novel functions and mechanisms may be uncovered by studying rare GAG residues/sequences.
Co-reporter:Akul Y. Mehta ; Jay N. Thakkar ; Bassem M. Mohammed ; Erika J. Martin ; Donald F. Brophy ; Takao Kishimoto ;Umesh R. Desai
Journal of Medicinal Chemistry 2014 Volume 57(Issue 7) pp:3030-3039
Publication Date(Web):March 17, 2014
DOI:10.1021/jm4020026
Exosite 2 of human thrombin contributes to two opposing pathways, the anticoagulant pathway and the platelet aggregation pathway. We reasoned that an exosite 2 directed allosteric thrombin inhibitor should simultaneously induce anticoagulant and antiplatelet effects. To assess this, we synthesized SbO4L based on the sulfated tyrosine-containing sequence of GPIbα. SbO4L was synthesized in three simple steps in high yield and found to be a highly selective, direct inhibitor of thrombin. Michelis–Menten kinetic studies indicated a noncompetitive mechanism of inhibition. Competitive inhibition studies suggested ideal competition with heparin and glycoprotein Ibα, as predicted. Studies with site-directed mutants of thrombin indicated that SbO4L binds to Arg233, Lys235, and Lys236 of exosite 2. SbO4L prevented thrombin-mediated platelet activation and aggregation as expected on the basis of competition with GPIbα. SbO4L presents a novel paradigm of simultaneous dual anticoagulant and antiplatelet effects achieved through the GPIbα binding site of thrombin.
Co-reporter:Malaika D. Argade ; Akul Y. Mehta ; Aurijit Sarkar ;Umesh R. Desai
Journal of Medicinal Chemistry 2014 Volume 57(Issue 8) pp:3559-3569
Publication Date(Web):March 25, 2014
DOI:10.1021/jm5002698
Factor XIa (fXIa) is being recognized as a prime target for developing safer anticoagulants. To discover synthetic, small, allosteric inhibitors of fXIa, we screened an in-house, unique library of 65 molecules displaying many distinct scaffolds and varying levels of sulfation. Of these, monosulfated benzofurans were the only group of molecules found to inhibit fXIa (∼100% efficacy) and led to the identification of monosulfated trimer 24 (IC50 0.82 μM) as the most potent inhibitor. Michaelis–Menten kinetics studies revealed a classic noncompetitive mechanism of action for 24. Although monosulfated, the inhibitors did not compete with unfractionated heparin alluding to a novel site of interaction. Fluorescence quenching studies indicated that trimer 24 induces major conformational changes in the active site of fXIa. Docking studies identified a site near Lys255 on the A3 domain of fXIa as the most probable site of binding for 24. Factor XIa devoid of the A3 domain displayed a major defect in the inhibition potency of 24 supporting the docking prediction. Our work presents the sulfated benzofuran scaffold as a promising framework to develop allosteric fXIa inhibitors that likely function through the A3 domain.
Co-reporter:Rami A. Al-Horani ;Umesh R. Desai
Journal of Medicinal Chemistry 2014 Volume 57(Issue 11) pp:4805-4818
Publication Date(Web):May 20, 2014
DOI:10.1021/jm500311e
We recently introduced sulfated pentagalloylglucopyranoside (SPGG) as an allosteric inhibitor of factor XIa (FXIa) (Al-Horani et al., J. Med Chem. 2013, 56, 867–878). To better understand the SPGG–FXIa interaction, we utilized eight SPGG variants and a range of biochemical techniques. The results reveal that SPGG’s sulfation level moderately affected FXIa inhibition potency and selectivity over thrombin and factor Xa. Variation in the anomeric configuration did not affect potency. Interestingly, zymogen factor XI bound SPGG with high affinity, suggesting its possible use as an antidote. Acrylamide quenching experiments suggested that SPGG induced significant conformational changes in the active site of FXIa. Inhibition studies in the presence of heparin showed marginal competition with highly sulfated SPGG variants but robust competition with less sulfated variants. Resolution of energetic contributions revealed that nonionic forces contribute nearly 87% of binding energy suggesting a strong possibility of specific interaction. Overall, the results indicate that SPGG may recognize more than one anion-binding, allosteric site on FXIa. An SPGG molecule containing approximately 10 sulfate groups on positions 2 through 6 of the pentagalloylglucopyranosyl scaffold may be the optimal FXIa inhibitor for further preclinical studies.
Co-reporter:Nirmita J. Patel, Rajesh Karuturi, Rami A. Al-Horani, Somesh Baranwal, Jagrut Patel, Umesh R. Desai, and Bhaumik B. Patel
ACS Chemical Biology 2014 Volume 9(Issue 8) pp:1826
Publication Date(Web):June 10, 2014
DOI:10.1021/cb500402f
Selective targeting of cancer stem-like cells (CSCs) is a paradigm-shifting approach. We hypothesized that CSCs can be targeted by interfering with functions of sulfated glycosaminoglycans, which play key roles in cancer cell growth, invasion and metastasis. We developed a tandem, dual screen strategy involving (1) assessing inhibition of monolayer versus spheroid growth and (2) assessing inhibition of primary versus secondary spheroid growth to identify G2.2, a unique sulfated nonsaccharide GAG mimetic (NSGM) from a focused library of 53 molecules, as a selective inhibitor of colon CSCs. The NSGM down-regulated several CSC markers through regulation of gene transcription, while closely related, inactive NSGMs G1.4 and G4.1 demonstrated no such changes. G2.2’s effects on CSCs were mediated, in part, through induction of apoptosis and inhibition of self-renewal factors. Overall, this work presents the proof-of-principle that CSCs can be selectively targeted through novel NSGMs, which are likely to advance fundamental understanding on CSCs while also aiding development of novel therapeutic agents.
Co-reporter:Preetpal Singh Sidhu ; May H. Abdel Aziz ; Aurijit Sarkar ; Akul Y. Mehta ; Qibing Zhou ;Umesh R. Desai
Journal of Medicinal Chemistry 2013 Volume 56(Issue 12) pp:5059-5070
Publication Date(Web):May 29, 2013
DOI:10.1021/jm400369q
We recently designed a group of novel exosite-2-directed sulfated, small, allosteric inhibitors of thrombin. To develop more potent inhibitors, monosulfated benzofuran tri- and tetrameric homologues of the parent designed dimers were synthesized in seven to eight steps and found to exhibit a wide range of potencies. Among these, trimer 9a was found to be nearly 10-fold more potent than the first generation molecules. Michaelis–Menten studies indicated an allosteric mechanism of inhibition. Competitive studies using a hirudin peptide (exosite 1 ligand) and unfractionated heparin, heparin octasaccharide, and γ′-fibrinogen peptide (exosite 2 ligands) demonstrated exosite 2 recognition in a manner different from that of the parent dimers. Alanine scanning mutagenesis of 12 Arg/Lys residues of exosite 2 revealed a defect in 9a potency for Arg233Ala thrombin only confirming the major difference in site of recognition between the two structurally related sulfated benzofurans. The results suggest that multiple avenues are available within exosite 2 for inducing thrombin inhibition.
Co-reporter:Rami A. Al-Horani ; Pooja Ponnusamy ; Akul Y. Mehta ; David Gailani ;Umesh R. Desai
Journal of Medicinal Chemistry 2013 Volume 56(Issue 3) pp:867-878
Publication Date(Web):January 14, 2013
DOI:10.1021/jm301338q
Inhibition of factor XIa (FXIa) is a novel paradigm for developing anticoagulants without major bleeding consequences. We present the discovery of sulfated pentagalloylglucoside (6) as a highly selective inhibitor of human FXIa. Biochemical screening of a focused library led to the identification of 6, a sulfated aromatic mimetic of heparin. Inhibitor 6 displayed a potency of 551 nM against FXIa, which was at least 200-fold more selective than other relevant enzymes. It also prevented activation of factor IX and prolonged human plasma and whole blood clotting. Inhibitor 6 reduced VMAX of FXIa hydrolysis of chromogenic substrate without affecting the KM, suggesting an allosteric mechanism. Competitive studies showed that 6 bound in the heparin-binding site of FXIa. No allosteric small molecule has been discovered to date that exhibits equivalent potency against FXIa. Inhibitor 6 is expected to open up a major route to allosteric FXIa anticoagulants with clinical relevance.
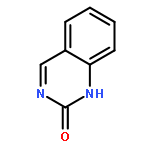
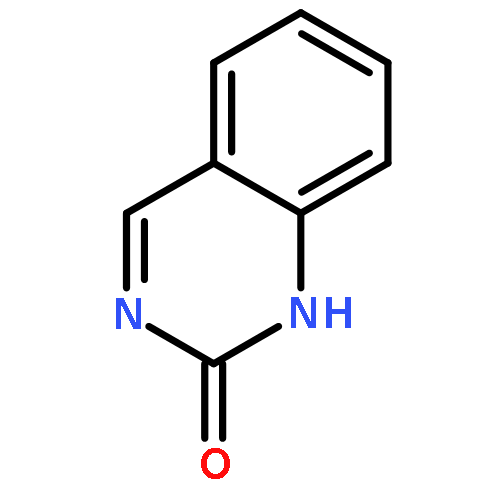
Co-reporter:Rajesh Karuturi ; Rami A. Al-Horani ; Shrenik C. Mehta ; David Gailani ;Umesh R. Desai
Journal of Medicinal Chemistry 2013 Volume 56(Issue 6) pp:2415-2428
Publication Date(Web):March 1, 2013
DOI:10.1021/jm301757v
To discover promising sulfated allosteric modulators (SAMs) of glycosaminoglycan-binding proteins (GBPs), such as human factor XIa (FXIa), we screened a library of 26 synthetic, sulfated quinazolin-4(3H)-ones (QAOs) resulting in the identification of six molecules that reduced the Vmax of substrate hydrolysis without influencing the KM. Mutagenesis of residues of the heparin-binding site (HBS) of FXIa introduced a nearly 5-fold loss in inhibition potency supporting recognition of an allosteric site. Fluorescence studies showed a sigmoidal binding profile indicating highly cooperative binding. Competition with a positively charged, heparin-binding polymer did not fully nullify inhibition suggesting importance of hydrophobic forces to binding. This discovery suggests the operation of a dual-element recognition process, which relies on an initial Coulombic attraction of anionic SAMs to the cationic HBS of FXIa that forms a locked complex through tight interaction with an adjacent hydrophobic patch. The dual-element strategy may be widely applicable for discovering SAMs of other GBPs.
Co-reporter:Preetpal S. Sidhu, Philip D. Mosier, Qibing Zhou, Umesh R. Desai
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 1) pp:355-359
Publication Date(Web):1 January 2013
DOI:10.1016/j.bmcl.2012.10.079
The design of sulfated, small, nonsaccharide molecules as modulators of proteins is still in its infancy as standard drug discovery tools such as library of diverse sulfated molecules and in silico docking and scoring protocol have not been firmly established. Databases, such as ZINC, contain too few sulfate-containing nonsaccharide molecules, which severely limits the identification of new hits. Lack of a generally applicable protocol for scaffold hopping limits the development of sulfated small molecules as synthetic mimetics of the highly sulfated glycosaminoglycans. We explored a sequential ligand-based (LBVS) and structure-based virtual screening (SBVS) approach starting from our initial discovery of monosulfated benzofurans to discover alternative scaffolds as allosteric modulators of thrombin, a key coagulation enzyme. Screening the ZINC database containing nearly 1 million nonsulfated small molecules using a pharmacophore developed from the parent sulfated benzofurans followed by a genetic algorithm-based dual-filter docking and scoring screening identified a group of 10 promising hits, of which three top-scoring hits were synthesized. Each was found to selectively inhibit human alpha-thrombin suggesting the possibility of this approach for scaffold hopping. Michaelis–Menten kinetics showed allosteric inhibition mechanism for the best molecule and human plasma studies confirmed good anticoagulation potential as expected. Our simple sequential LBVS and SBVS approach is likely to be useful as a general strategy for identification of sulfated small molecules hits as modulators of glycosaminoglycan–protein interactions.
Co-reporter:May H. Abdel Aziz ; Preetpal Singh Sidhu ; Aiye Liang ; Ji Yeong Kim ; Philip D. Mosier ; Qibing Zhou ; David H. Farrell ;Umesh R. Desai
Journal of Medicinal Chemistry 2012 Volume 55(Issue 15) pp:6888-6897
Publication Date(Web):July 12, 2012
DOI:10.1021/jm300670q
Earlier, we reported on the design of sulfated benzofuran dimers (SBDs) as allosteric inhibitors of thrombin (Sidhu et al. J. Med. Chem.201154 5522–5531). To identify the site of binding of SBDs, we studied thrombin inhibition in the presence of exosite 1 and 2 ligands. Whereas hirudin peptide and heparin octasaccharide did not affect the IC50 of thrombin inhibition by a high affinity SBD, the presence of full-length heparin reduced inhibition potency by 4-fold. The presence of γ′ fibrinogen peptide, which recognizes Arg93, Arg97, Arg173, Arg175, and other residues, resulted in a loss of affinity that correlated with the ideal Dixon–Webb competitive profile. Replacement of several arginines and lysines of exosite 2 with alanine did not affect thrombin inhibition potency, except for Arg173, which displayed a 22-fold reduction in IC50. Docking studies suggested a hydrophobic patch around Arg173 as a plausible site of SBD binding to thrombin. The absence of the Arg173-like residue in factor Xa supported the observed selectivity of inhibition by SBDs. Cellular toxicity studies indicated that SBDs are essentially nontoxic to cells at concentrations as high as 250 mg/kg. Overall, the work presents the localization of the SBD binding site, which could lead to allosteric modulators of thrombin that are completely different from all clinically used anticoagulants.
Co-reporter:Rami A. Al-Horani, Umesh R. Desai
Tetrahedron 2012 68(8) pp: 2027-2040
Publication Date(Web):
DOI:10.1016/j.tet.2012.01.005
Co-reporter:Preetpal Singh Sidhu ; Aiye Liang ; Akul Y. Mehta ; May H. Abdel Aziz ; Qibing Zhou ;Umesh R. Desai
Journal of Medicinal Chemistry 2011 Volume 54(Issue 15) pp:5522-5531
Publication Date(Web):June 29, 2011
DOI:10.1021/jm2005767
Thrombin is a key enzyme targeted by the majority of current anticoagulants that are direct inhibitors. Allosteric inhibition of thrombin may offer a major advantage of finely tuned regulation. We present here sulfated benzofurans as the first examples of potent, small allosteric inhibitors of thrombin. A sulfated benzofuran library of 15 sulfated monomers and 13 sulfated dimers with different charged, polar, and hydrophobic substituents was studied in this work. Synthesis of the sulfated benzofurans was achieved through a multiple step, highly branched strategy, which culminated with microwave-assisted chemical sulfation. Of the 28 potential inhibitors, 11 exhibited reasonable inhibition of human α-thrombin at pH 7.4. Structure–activity relationship analysis indicated that sulfation at the 5-position of the benzofuran scaffold was essential for targeting thrombin. A tert-butyl 5-sulfated benzofuran derivative was found to be the most potent thrombin inhibitor with an IC50 of 7.3 μM under physiologically relevant conditions. Michaelis–Menten studies showed an allosteric inhibition phenomenon. Plasma clotting assays indicate that the sulfated benzofurans prolong both the activated partial thromboplastin time and prothrombin time. Overall, this work puts forward sulfated benzofurans as the first small, synthetic molecules as powerful lead compounds for the design of a new class of allosteric inhibitors of thrombin.
Co-reporter:Aiye Liang;Jay N. Thakkar ;Umesh R. Desai
Journal of Pharmaceutical Sciences 2010 Volume 99( Issue 3) pp:1207-1216
Publication Date(Web):
DOI:10.1002/jps.21908
Abstract
Heparin (H) and heparan sulfate (HS) play major roles in a number of biological processes. Yet, H/HS-based pharmaceutical agents are also associated with multiple adverse effects. This has led to the concept of designing noncarbohydrate, aromatic mimetics that modulate H/HS function. In this work, we study a library of synthetic, aromatic H/HS mimetics for their capillary electrophoretic profiles, the acid and base stability, and aqueous–organic partitioning property. The nonsugar H/HS mimetics exhibit electrophoretic properties similar to sulfated oligosaccharides suggesting that the mimetics can be rapidly and quantitatively analyzed. Stability studies show that the mimetics are essentially stable under neutral and basic conditions in a manner similar to the heparins, but are considerably unstable under acidic conditions in contrast to heparins. The measurement of partition coefficients show major differences within the sulfated mimetics as well as between the measured and calculated log P values. Understanding these physico-chemical properties is expected to have significant implications in the pharmaceutical development of this growing class of molecules. © 2009 Wiley-Liss, Inc. and the American Pharmacists Association J Pharm Sci 99: 1207–1216, 2010
Co-reporter:Arjun Raghuraman, Philip D. Mosier and Umesh R. Desai
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 6) pp:281
Publication Date(Web):June 14, 2010
DOI:10.1021/ml100048y
Dermatan sulfate, an important member of the glycosaminoglycan family, interacts with heparin cofactor II, a member of the serpin family of proteins, to modulate antithrombotic response. Yet, the nature of this interaction remains poorly understood at a molecular level. We report the genetic algorithm-based combinatorial virtual library screening study of a natural, high-affinity dermatan sulfate hexasaccharide with heparin cofactor II. Of the 192 topologies possible for the hexasaccharide, only 16 satisfied the “high-specificity” criteria used in computational study. Of these, 13 topologies were predicted to bind in the heparin-binding site of heparin cofactor II at a ∼60° angle to helix D, a novel binding mode. This new binding geometry satisfies all known solution and mutagenesis data and supports thrombin ternary complexation through a template mechanism. The study is expected to facilitate the design of allosteric agonists of heparin cofactor II as antithrombotic agents.Keywords (KEYWORDS): Computational biology; dermatan sulfate; glycosaminoglycans; heparin cofactor II; serpins; structure−activity relationships
Co-reporter:Rami A. Al-Horani, Umesh R. Desai
Tetrahedron 2010 66(16) pp: 2907-2918
Publication Date(Web):
DOI:10.1016/j.tet.2010.02.015
Co-reporter:Jay N. Thakkar, Vaibhav Tiwari and Umesh R. Desai
Biomacromolecules 2010 Volume 11(Issue 5) pp:
Publication Date(Web):April 22, 2010
DOI:10.1021/bm100161u
In an effort to discover macromolecular mimetics of heparan sulfate (HS), we previously designed sulfated lignins (Raghuraman et al. Biomacromolecules 2007, 8, 1759−1763). To probe the relevance of sulfate groups of HS in viral entry, lignins completely devoid of sulfate moieties, and yet possessing an electrostatic surface equivalent to that of HS, were designed. Two carboxylated lignins based on a 4-hydroxy cinnamic acid scaffold were synthesized using enzymatic oxidative coupling in high yields, fractionated according to their sizes, and tested in cellular assays of herpes simplex virus-1 (HSV-1) infection. The two carboxylated lignins were found to not only inhibit HSV-1 entry into mammalian cells (IC50 = 8−56 nM), but were more potent than sulfated lignins. In addition, shorter carboxylated lignins were found to be as active as the longer chains, suggesting that structural features, in addition to carboxylate groups, may be important. It can be expected that carboxylated lignins also antagonize the entry of other enveloped viruses, for example, HIV-1, Kaposi’s sarcoma-associated herpes virus, and hepatitis C virus, that utilize HS to gain entry into cells. The results present major opportunities for developing lignin-based antiviral formulations for topical use.
Co-reporter:Arjun Raghuraman, Aiye Liang, Chandravel Krishnasamy, Trish Lauck, Gunnar T. Gunnarsson, Umesh R. Desai
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 6) pp:2626-2631
Publication Date(Web):June 2009
DOI:10.1016/j.ejmech.2008.09.042
Antithrombin, a plasma glycoprotein serpin, requires conformational activation by heparin to induce an anticoagulant effect, which is mediated through accelerated factor Xa inhibition. Heparin, a highly charged polymer and an allosteric activator of the serpin, is associated with major adverse effects. To design better, but radically different activators of antithrombin from heparin, we utilized a pharmacophore-based approach. A tetrahydroisoquinoline-based scaffold was designed to mimic four critical anionic groups of the key trisaccharide DEF constituting the sequence-specific pentasaccharide DEFGH in heparin. Activator IAS5 containing 5,6-disulfated tetrahydroisoquinoline and 3,4,5-trisulfated phenyl rings was found to bind antithrombin at pH 7.4 with an affinity comparable to the reference trisaccharide DEF. IAS5 activated the inhibitor nearly 30-fold, nearly 2- to 3-fold higher than our first generation flavanoid-based designs. This work advances the concept of antithrombin activation through non-saccharide, organic molecules and pinpoints a direction for the design of more potent molecules.
Co-reporter:Jenson Verghese, Aiye Liang, Preet Pal Singh Sidhu, Michael Hindle, Qibing Zhou, Umesh R. Desai
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 15) pp:4126-4129
Publication Date(Web):1 August 2009
DOI:10.1016/j.bmcl.2009.06.013
Designing non-saccharide functional mimics of heparin is a major challenge. In this work, a library of small, aromatic molecules based on the sulfated DHP scaffold was synthesized and screened against thrombin and factor Xa. The results reveal that (i) selected monomeric benzofuran derivatives inhibit the two enzymes, albeit weakly; (ii) the two enzymes recognize different structural features in the benzofurans studied suggesting significant selectivity of recognition; and (iii) the mechanism of inhibition is allosteric. The molecules represent the first allosteric small molecule inhibitors of the two enzymes.
Co-reporter:Chravel Krishnasamy;Arjun Raghuraman;LemontB. Kier;UmeshR. Desai
Chemistry & Biodiversity 2008 Volume 5( Issue 12) pp:2609-2620
Publication Date(Web):
DOI:10.1002/cbdv.200890216
Abstract
Factor Xa and thrombin, two critical pro-coagulant enzymes of the clotting cascade, are the primary target of current anticoagulation research that aims to develop potent, orally bioavailable, synthetic small-molecule inhibitors. To determine structural features that might play important roles in factor Xa and thrombin recognition and oral bioavailability, quantitative structure–activity and structure–property analyses were performed on the factor Xa and thrombin inhibition data and Caco-2 cell-permeability data of 3-substituted pyrazole-5-carboxamides reported by Pinto et al. (J. Med. Chem.2001, 44, 566). The factor Xa and thrombin inhibition potencies, and Caco-2 cell permeability of the 3-substituted pyrazole-5-carboxamides could be quantitatively described through molecular connectivity and atom level E-state indices. Different quantitative structure–activity and structure–property models were derived for each of the three biological properties. The models are statistically relevant with correlation coefficients of at least 0.9, and contain only two or three molecular descriptor variables. The study demonstrates the use of molecular connectivity and E-state indices in understanding factor Xa and thrombin inhibition. In addition, the models may be useful for predictive purposes in generating molecules with better potency, specificity, and oral bioavailability.
Co-reporter:Mohammed Rahman;Muhammad Riaz;Umesh R. Desai
Chemistry & Biodiversity 2007 Volume 4(Issue 11) pp:2495-2527
Publication Date(Web):16 NOV 2007
DOI:10.1002/cbdv.200790205
Recent investigations show that naturally occurring biflavanoids possess anti-inflammatory, anticancer, antiviral, antimicrobial, vasorelaxant, and anticlotting activities. These activities have been discovered from the small number of biflavanoid structures that have been investigated, although the natural biflavanoid library is likely to be large. Structurally, biflavanoids are polyphenolic molecules comprised of two identical or non-identical flavanoid units conjoined in a symmetrical or unsymmetrical manner through an alkyl or an alkoxy-based linker of varying length. These possibilities introduce significant structural variation in biflavanoids, which is further amplified by the positions of functional groups – hydroxy, methoxy, keto, or double bond – and stereogenic centers on the flavanoid scaffold. In combination, the class of biflavanoids represents a library of structurally diverse molecules, which remains to be fully exploited. Since the time of their discovery, several chemical approaches utilizing coupling and rearrangement strategies have been developed to synthesize biflavanoids. This review compiles these synthetic approaches into nine different methods including Ullmann coupling of halogenated flavones, biphenyl-based construction of biflavanoids, metal-catalyzed cross-coupling of flavones, Wessely–Moser rearrangements, oxidative coupling of flavones, Ullmann condensation with nucleophiles, nucleophilic substitutions for alkoxy biflavanoids, and dehydrogenation-based or hydrogenation-based synthesis. Newer, more robust synthetic approaches are necessary to realize the full potential of the structurally diverse class of biflavanoids.
Co-reporter:Mandakini Dantuluri, Gunnar T. Gunnarsson, Muhammad Riaz, Huyen Nguyen, Umesh R. Desai
Analytical Biochemistry 2005 Volume 336(Issue 2) pp:316-322
Publication Date(Web):15 January 2005
DOI:10.1016/j.ab.2004.10.017
Flavanoids and flavonoids are natural products present in our diet and known to possess multiple biological activities. Sulfated species of these natural products represent highly charged water-soluble organic molecules that possess unique biochemical properties. We describe here the first studies on capillary electrophoresis of these highly charged molecules. Fully sulfated flavanoids and flavonoids can be electrophoresed and resolved under reverse polarity at pH 3.5 using 5–10 kV in less than 20 min. In contrast, at high pH under normal polarity these species can be electrophoresed only if a pressurized capillary is employed. (±)-Catechin sulfate, a racemic sulfated flavanoid, was resolved into its enantiomers using 15% β-cyclodextrin, a chiral selector, but not with α- or γ-cyclodextrins. Yet, the high charge density of these molecules challenges the resolving capability of capillary electrophoresis as diastereomers (−)-epicatechin sulfate and (+)-catechin sulfate do not resolve, even in the presence of cyclodextrins or chiral positively charged amino acids. Overall, capillary electrophoresis of highly sulfated flavanoids and flavonoids is expected to be useful in rapid structure analysis of sulfated flavonoids, either synthetic or natural.
Co-reporter:Bhawana Saluja, Jay N. Thakkar, Hua Li, Umesh R. Desai, Masahiro Sakagami
Pulmonary Pharmacology & Therapeutics (April 2013) Volume 26(Issue 2) pp:296-304
Publication Date(Web):1 April 2013
DOI:10.1016/j.pupt.2012.12.009
No molecule has been found to be effective against emphysema to date primarily because of its complex pathogenesis that involves elastolysis, oxidation and inflammation. We here describe novel unsulfated or sulfated low molecular weight lignins (LMWLs) chemo-enzymatically prepared from 4-hydroxycinnamic acids monomers, as the first potent triple-action inhibitors of neutrophil elastase, oxidation and inflammation. The inhibitory potencies of three different cinnamic acid-based LMWLs were determined in vitro using chromogenic substrate hydrolysis assays, radical scavenging and lung cellular oxidative biomarker reduced glutathione (rGSH) assays, and lung cellular inflammatory biomarker NFκB and IL-8 assays, respectively. Each LWML uniquely displayed triple-action inhibition, among which CDSO3, a sulfated caffeic acid-based LMWL, was most potent. The half-maximal anti-human neutrophil elastase (HNE) potency of CDSO3 was 0.43 μM. This high potency arose from lignin-like oligomerization, which was further potentiated by 6.6-fold due to sulfation. Mechanistically, this elastase inhibition was of mixed-type, time-dependent and more selective to positively charged elastases. The half-maximal anti-oxidative potency of CDSO3 was 3.52 μM, 4.8-fold potentiated from that of the monomer, caffeic acid (CA). In contrast, the half-maximal inhibitory potency to TNFα-induced inflammation was 5–10 μM, despite no activity with the monomer. More intriguingly, this anti-inflammatory activity was essentially identical with different stimuli, okadaic acid and hydrogen peroxide (H2O2), which implied that CDSO3 acts directly on inflammatory cascades within the cells. Overall, oligomerization and sulfation produced or significantly potentiated the activity, in comparison to the monomer. Thus, sulfated and unsulfated LMWLs are novel non-peptidic 2.8–4.1 kDa macromolecules that exhibit for the first time potent triple inhibitory activity against elastase, oxidation and inflammation, the three major pathogenic mechanisms known to cause emphysema.
Co-reporter:J. Timothy King, Umesh R. Desai
Analytical Biochemistry (15 September 2008) Volume 380(Issue 2) pp:
Publication Date(Web):15 September 2008
DOI:10.1016/j.ab.2008.05.046
Clinically used low molecular weight heparins (LMWH) are anticoagulants of choice and are phenomenally complex mixtures of millions of distinct natural and unnatural polymeric sequences. The FDA recommends that each LMWH be considered as an independent drug with its own activity profile, placing significant importance on the biophysical characterization of each intact LMWH. We report a robust protocol for fingerprinting these pharmaceutical agents. Capillary electrophoresis of three LMWHs, enoxaparin, tinzaparin, and a Sigma preparation, under reverse polarity conditions in the presence of selected linear alkyl polyamines gives an electrophoretic pattern that is characteristic of the nature of the starting material. The buffers that best provided optimal resolution without compromising sensitivity and speed of analysis were 50 mM sodium phosphate, pH 2.3, and 100 mM ammonium formate, pH 3.5. Resolution was strongly dependent on the structure of polyamine with pentaethylenehexamine being most effective for enoxaparin and Sigma LMWH. In contrast, tinzaparin could be best resolved with tetraethylenepentamine. Cyclic polyamines were ineffective. Resolution was also dependent on the concentration of resolving agents and displayed a narrow window that provides optimal resolution. These features suggest a strong structural origin of the fingerprint pattern. Overall, the simple protocol will find special use in assessing LMWH quality and batch-to-batch variability.
Co-reporter:Rami A. Al-Horani ; Aiye Liang ;Umesh R. Desai
Journal of Medicinal Chemistry () pp:
Publication Date(Web):July 29, 2011
DOI:10.1021/jm2008387
Antithrombin is a key regulator of coagulation and prime target of heparins, clinically used anticoagulants. Heparins induce a two-step conformational activation of antithrombin, a process that has remained challenging to target with molecules devoid of the antithrombin-binding pentasaccharide DEFGH. Computational screening of a focused library led to the design of two tetra-sulfated N-arylacyl tetrahydroisoquinoline variants as potential nonsaccharide activators of antithrombin. A high yielding synthetic scheme based on Horner–Wadsworth–Emmons or Pictet–Spengler reactions was developed to facilitate the functionalization of the tetrahydoisoquinoline ring, which upon further amidation, deprotection, and sulfation gave the targeted nonsaccharide activators. Spectrofluorometric measurement of affinity displayed antithrombin binding affinities in the low to high micromolar range at pH 6.0, I 0.05, 25 °C. Measurement of second-order rate constants of antithrombin inhibition of factor Xa in the presence and absence of the designed activators showed antithrombin activation in the range of 8–80-fold in the pH 6.0 buffer. This work puts forward 20c, a novel tetra-sulfated N-arylacyl tetrahydroisoquinoline-based molecule, that activates AT only 3.8-fold less than that achieved with DEFGH, suggesting a strong possibility of rationally designing sulfated organic molecules as clinically relevant AT activators.










![b-D-Glucopyranose,pentakis[3,4,5-tris(phenylmethoxy)benzoate] (9CI)](http://img.cochemist.com/ccimg/122700/122625-60-9.png)
![b-D-Glucopyranose,pentakis[3,4,5-tris(phenylmethoxy)benzoate] (9CI)](http://img.cochemist.com/ccimg/122700/122625-60-9_b.png)









![[(2r,3r,4s,5r,6s)-3,4,5,6-tetrakis[(3,4,5-trihydroxybenzoyl)oxy]oxan-2-yl]methyl 3,4,5-trihydroxybenzoate](http://img.cochemist.com/ccimg/15000/14937-32-7.png)
![[(2r,3r,4s,5r,6s)-3,4,5,6-tetrakis[(3,4,5-trihydroxybenzoyl)oxy]oxan-2-yl]methyl 3,4,5-trihydroxybenzoate](http://img.cochemist.com/ccimg/15000/14937-32-7_b.png)