Co-reporter:Rajesh K. Singh, Yaniv Kazansky, Donald Wathieu, and David Fushman
Analytical Chemistry August 1, 2017 Volume 89(Issue 15) pp:7852-7852
Publication Date(Web):July 7, 2017
DOI:10.1021/acs.analchem.6b04194
Protein ubiquitination plays a role in essentially every process in eukaryotic cells. The attachment of ubiquitin (Ub) or Ub-like (UBL) proteins to target proteins is achieved by parallel but distinct cascades of enzymatic reactions involving three enzymes: E1, E2, and E3. The E1 enzyme functions at the apex of this pathway and plays a critical role in activating the C-terminus of ubiquitin or UBL, which is an essential step that triggers subsequent downstream transfer to their cognate E2s resulting in the fidelity of the Ub/UBL conjugation machinery. Despite the central role of the E1 enzyme in protein modification, a quantitative method to measure Ub/UBL activation by E1 is lacking. Here, we present a mass spectrometry-based assay to accurately measure the activation of Ub/UBL by E1 independent of the E2/E3 enzymes. Our method does not require radiolabeling of any components and therefore can be used in any biochemical laboratory having access to a mass spectrometer. This method allowed us to dissect the concerted process of E1-E2-catalyzed Ub conjugation in order to separately characterize the process of Ub activation and how it is affected by select mutations and other factors. We found that the hydrophobic patch of Ub is important for the optimal activation of Ub by E1. We further show that the blockers of the Ub-proteasome system such as ubistatin and fullerenol inhibit Ub activation by E1. Interestingly, our data indicate that the phosphorylation of Ub at the S65 position augments its activation by the E1 enzyme.
Co-reporter:Michal Chojnacki, Wissam Mansour, Dharjath S. Hameed, Rajesh K. Singh, ... Michael H. Glickman
Cell Chemical Biology 2017 Volume 24, Issue 4(Volume 24, Issue 4) pp:
Publication Date(Web):20 April 2017
DOI:10.1016/j.chembiol.2017.02.013
•Photoleucine was successfully incorporated into fully synthetic ubiquitin monomers•Embedded photoleucine permitted binding to the hydrophobic patch of ubiquitin•Enzymatically polymerized ubiquitin phototrap captured Ub-binding receptors•The first PC region of Rpn1, either isolated or proteasome-incorporated, bound polyUbUbiquitin (Ub) signaling is a diverse group of processes controlled by covalent attachment of small protein Ub and polyUb chains to a range of cellular protein targets. The best documented Ub signaling pathway is the one that delivers polyUb proteins to the 26S proteasome for degradation. However, studies of molecular interactions involved in this process have been hampered by the transient and hydrophobic nature of these interactions and the lack of tools to study them. Here, we develop Ub-phototrap (UbPT), a synthetic Ub variant containing a photoactivatable crosslinking side chain. Enzymatic polymerization into chains of defined lengths and linkage types provided a set of reagents that led to identification of Rpn1 as a third proteasome ubiquitin-associating subunit that coordinates docking of substrate shuttles, unloading of substrates, and anchoring of polyUb conjugates. Our work demonstrates the value of UbPT, and we expect that its future uses will help define and investigate the ubiquitin interactome.Download high-res image (137KB)Download full-size image
Co-reporter:Mark A. Nakasone, Timothy A. Lewis, Olivier Walker, Anita Thakur, ... David Fushman
Structure 2017 Volume 25, Issue 12(Volume 25, Issue 12) pp:
Publication Date(Web):5 December 2017
DOI:10.1016/j.str.2017.10.007
•Ubistatin B binds Ub selectively and prefers K48-linked Ub chains over K11 or K63•Hydrophobic and charge/polar interactions are critical for ubistatin:Ub binding•Ubistatins block disassembly of Ub conjugates by various DUBs and by 26S proteasome•Ubistatin B penetrates cancer cells and alters the cellular Ub landscapeThe discovery of ubistatins, small molecules that impair proteasomal degradation of proteins by directly binding to polyubiquitin, makes ubiquitin itself a potential therapeutic target. Although ubistatins have the potential for drug development and clinical applications, the lack of structural details of ubiquitin-ubistatin interactions has impeded their development. Here, we characterized a panel of new ubistatin derivatives using functional and binding assays. The structures of ubiquitin complexes with ubistatin B and hemi-ubistatin revealed direct interactions with ubiquitin's hydrophobic surface patch and the basic/polar residues surrounding it. Ubistatin B binds ubiquitin and diubiquitin tighter than a high-affinity ubiquitin receptor and shows strong preference for K48 linkages over K11 and K63. Furthermore, ubistatin B shields ubiquitin conjugates from disassembly by a range of deubiquitinases and by the 26S proteasome. Finally, ubistatin B penetrates cancer cells and alters the cellular ubiquitin landscape. These findings highlight versatile properties of ubistatins and have implications for their future development and use in targeting ubiquitin-signaling pathways.Download high-res image (157KB)Download full-size image
Co-reporter:Carlos A. Castañeda, Apurva Chaturvedi, Christina M. Camara, Joseph E. Curtis, Susan Krueger and David Fushman
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 8) pp:5771-5788
Publication Date(Web):21 Sep 2015
DOI:10.1039/C5CP04601G
Polyubiquitination is a critical protein post-translational modification involved in a variety of processes in eukaryotic cells. The molecular basis for selective recognition of the polyubiquitin signals by cellular receptors is determined by the conformations polyubiquitin chains adopt; this has been demonstrated for K48- and K63-linked chains. Recent studies of the so-called non-canonical chains (linked via K6, K11, K27, K29, or K33) suggest they play important regulatory roles in growth, development, and immune system pathways, but biophysical studies are needed to elucidate the physical/structural basis of their interactions with receptors. A first step towards this goal is characterization of the conformations these chains adopt in solution. We assembled diubiquitins (Ub2) comprised of every lysine linkage. Using solution NMR measurements, small-angle neutron scattering (SANS), and in silico ensemble generation, we determined population-weighted conformational ensembles that shed light on the structure and dynamics of the non-canonical polyubiquitin chains. We found that polyubiquitin is conformationally heterogeneous, and each chain type exhibits unique conformational ensembles. For example, K6-Ub2 and K11-Ub2 (at physiological salt concentration) are in dynamic equilibrium between at least two conformers, where one exhibits a unique Ub/Ub interface, distinct from that observed in K48-Ub2 but similar to crystal structures of these chains. Conformers for K29-Ub2 and K33-Ub2 resemble recent crystal structures in the ligand-bound state. Remarkably, a number of diubiquitins adopt conformers similar to K48-Ub2 or K63-Ub2, suggesting potential overlap of biological function among different lysine linkages. These studies highlight the potential power of determining function from elucidation of conformational states.
Co-reporter:Dr. Rajesh K. Singh;Adithya Sundar ; David Fushman
Angewandte Chemie International Edition 2014 Volume 53( Issue 24) pp:6120-6125
Publication Date(Web):
DOI:10.1002/anie.201402642
Abstract
Uncovering the mechanisms that allow conjugates of ubiquitin (Ub) and/or Ub-like (UBL) proteins such as Rub1 to serve as distinct molecular signals requires the ability to make them with native connectivity and defined length and linkage composition. A novel, effective, and affordable strategy for controlled chemical assembly of fully natural UBL–Ub, Ub–UBL, and UBL–UBL conjugates from recombinant monomers is presented. Rubylation of Ub and Rub1 and ubiquitination of Rub1 was achieved without E2/E3 enzymes. New residue-specific information was obtained on the interdomain contacts in naturally-occurring K48-linked Rub1–Ub and Ub–Rub1, and K29-linked Rub1–Ub heterodimers, and their recognition by a K48-linkage-specific Ub receptor. The disassembly of these heterodimers by major deubiquitinating enzymes was examined and it was discovered that some deubiquitinases also possess derubylase activity. This unexpected result suggests possible crosstalk between Ub and Rub1/Nedd8 signaling pathways.
Co-reporter:Dr. Rajesh K. Singh;Adithya Sundar ; David Fushman
Angewandte Chemie 2014 Volume 126( Issue 24) pp:6234-6239
Publication Date(Web):
DOI:10.1002/ange.201402642
Abstract
Uncovering the mechanisms that allow conjugates of ubiquitin (Ub) and/or Ub-like (UBL) proteins such as Rub1 to serve as distinct molecular signals requires the ability to make them with native connectivity and defined length and linkage composition. A novel, effective, and affordable strategy for controlled chemical assembly of fully natural UBL–Ub, Ub–UBL, and UBL–UBL conjugates from recombinant monomers is presented. Rubylation of Ub and Rub1 and ubiquitination of Rub1 was achieved without E2/E3 enzymes. New residue-specific information was obtained on the interdomain contacts in naturally-occurring K48-linked Rub1–Ub and Ub–Rub1, and K29-linked Rub1–Ub heterodimers, and their recognition by a K48-linkage-specific Ub receptor. The disassembly of these heterodimers by major deubiquitinating enzymes was examined and it was discovered that some deubiquitinases also possess derubylase activity. This unexpected result suggests possible crosstalk between Ub and Rub1/Nedd8 signaling pathways.
Co-reporter:Konstantin Berlin ; Carlos A. Castañeda ; Dina Schneidman-Duhovny ; Andrej Sali ; Alfredo Nava-Tudela
Journal of the American Chemical Society 2013 Volume 135(Issue 44) pp:16595-16609
Publication Date(Web):October 4, 2013
DOI:10.1021/ja4083717
Structural analysis of proteins and nucleic acids is complicated by their inherent flexibility, conferred, for example, by linkers between their contiguous domains. Therefore, the macromolecule needs to be represented by an ensemble of conformations instead of a single conformation. Determining this ensemble is challenging because the experimental data are a convoluted average of contributions from multiple conformations. As the number of the ensemble degrees of freedom generally greatly exceeds the number of independent observables, directly deconvolving experimental data into a representative ensemble is an ill-posed problem. Recent developments in sparse approximations and compressive sensing have demonstrated that useful information can be recovered from underdetermined (ill-posed) systems of linear equations by using sparsity regularization. Inspired by these advances, we designed the Sparse Ensemble Selection (SES) method for recovering multiple conformations from a limited number of observations. SES is more general and accurate than previously published minimum-ensemble methods, and we use it to obtain representative conformational ensembles of Lys48-linked diubiquitin, characterized by the residual dipolar coupling data measured at several pH conditions. These representative ensembles are validated against NMR chemical shift perturbation data and compared to maximum-entropy results. The SES method reproduced and quantified the previously observed pH dependence of the major conformation of Lys48-linked diubiquitin, and revealed lesser-populated conformations that are preorganized for binding known diubiquitin receptors, thus providing insights into possible mechanisms of receptor recognition by polyubiquitin. SES is applicable to any experimental observables that can be expressed as a weighted linear combination of data for individual states.
Co-reporter:Emma K. Dixon, Carlos A. Castañeda, Tanuja R. Kashyap, Yan Wang, David Fushman
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 12) pp:3421-3429
Publication Date(Web):15 June 2013
DOI:10.1016/j.bmc.2013.02.052
Polymeric chains of a small protein ubiquitin are involved in regulation of nearly all vital processes in eukaryotic cells. Elucidating the signaling properties of polyubiquitin requires the ability to make these chains in vitro. In recent years, chemical and chemical–biology tools have been developed that produce fully natural isopeptide-linked polyUb chains with no need for linkage-specific ubiquitin-conjugating enzymes. These methods produced unbranched chains (in which no more than one lysine per ubiquitin is conjugated to another ubiquitin). Here we report a nonenzymatic method for the assembly of fully natural isopeptide-linked branched polyubiquitin chains. This method is based on the use of mutually orthogonal removable protecting groups (e.g., Boc- and Alloc-) on lysines combined with an Ag-catalyzed condensation reaction between a C-terminal thioester on one ubiquitin and a specific ε-amine on another ubiquitin, and involves genetic incorporation of more than one Lys(Boc) at the desired linkage positions in the ubiquitin sequence. We demonstrate our method by making a fully natural branched tri-ubiquitin containing isopeptide linkages via Lys11 and Lys33, and a 15N-enriched proximal ubiquitin, which enabled monomer-specific structural and dynamical studies by NMR. Furthermore, we assayed disassembly of branched and unbranched tri-ubiquitins as well as control di-ubiquitins by the yeast proteasome-associated deubiquitinase Ubp6. Our results show that Ubp6 can recognize and disassemble a branched polyubiquitin, wherein cleavage preferences for individual linkages are retained. Our spectroscopic and functional data suggest that, at least for the chains studied here, the isopeptide linkages are effectively independent of each other. Together with our method for nonenzymatic assembly of unbranched polyubiquitin, these developments now provide tools for making fully natural polyubiquitin chains of essentially any type of linkage and length.
Co-reporter:Carlos Castañeda ; Jia Liu ; Apurva Chaturvedi ; Urszula Nowicka ; T. Ashton Cropp
Journal of the American Chemical Society 2011 Volume 133(Issue 44) pp:17855-17868
Publication Date(Web):September 30, 2011
DOI:10.1021/ja207220g
Polymeric chains made of a small protein ubiquitin act as molecular signals regulating a variety of cellular processes controlling essentially all aspects of eukaryotic biology. Uncovering the mechanisms that allow differently linked polyubiquitin chains to serve as distinct molecular signals requires the ability to make these chains with the native connectivity, defined length, linkage composition, and in sufficient quantities. This, however, has been a major impediment in the ubiquitin field. Here, we present a robust, efficient, and widely accessible method for controlled iterative nonenzymatic assembly of polyubiquitin chains using recombinant ubiquitin monomers as the primary building blocks. This method uses silver-mediated condensation reaction between the C-terminal thioester of one ubiquitin and the ε-amine of a specific lysine on the other ubiquitin. We augment the nonenzymatic approaches developed recently by using removable orthogonal amine-protecting groups, Alloc and Boc. The use of bacterially expressed ubiquitins allows cost-effective isotopic enrichment of any individual monomer in the chain. We demonstrate that our method yields completely natural polyubiquitin chains (free of mutations and linked through native isopeptide bonds) of essentially any desired length, linkage composition, and isotopic labeling scheme, and in milligram quantities. Specifically, we successfully made Lys11-linked di-, tri-, and tetra-ubiquitins, Lys33-linked diubiquitin, and a mixed-linkage Lys33,Lys11-linked triubiquitin. We also demonstrate the ability to obtain, by high-resolution NMR, residue-specific information on ubiquitin units at any desired position in such chains. This method opens up essentially endless possibilities for rigorous structural and functional studies of polyubiquitin signals.
Co-reporter:Dr. Carlos A. Castañeda;Liat Spasser;Dr. Sudhir N. Bavikar; Ashraf Brik; David Fushman
Angewandte Chemie International Edition 2011 Volume 50( Issue 47) pp:11210-11214
Publication Date(Web):
DOI:10.1002/anie.201104649
Co-reporter:Konstantin Berlin ; Dianne P. O’Leary
Journal of the American Chemical Society 2010 Volume 132(Issue 26) pp:8961-8972
Publication Date(Web):June 15, 2010
DOI:10.1021/ja100447p
We present and evaluate a rigid-body molecular docking method, called PATIDOCK, that relies solely on the three-dimensional structure of the individual components and the experimentally derived residual dipolar couplings (RDCs) for the complex. We show that, given an accurate ab initio predictor of the alignment tensor from a protein structure, it is possible to accurately assemble a protein−protein complex by utilizing the RDCs’ sensitivity to molecular shape to guide the docking. The proposed docking method is robust against experimental errors in the RDCs and computationally efficient. We analyze the accuracy and efficiency of this method using experimental or synthetic RDC data for several proteins, as well as synthetic data for a large variety of protein−protein complexes. We also test our method on two protein systems for which the structure of the complex and steric-alignment data are available (Lys48-linked diubiquitin and a complex of ubiquitin and a ubiquitin-associated domain) and analyze the effect of flexible unstructured tails on the outcome of docking. The results demonstrate that it is fundamentally possible to assemble a protein−protein complex solely on the basis of experimental RDC data and the prediction of the alignment tensor from 3D structures. Thus, despite the purely angular nature of RDCs, they can be converted into intermolecular distance/translational constraints. Additionally, we show a method for combining RDCs with other experimental data, such as ambiguous constraints from interface mapping, to further improve structure characterization of protein complexes.
Co-reporter:Konstantin Berlin, Dianne P. O’Leary, David Fushman
Journal of Magnetic Resonance 2009 Volume 201(Issue 1) pp:25-33
Publication Date(Web):November 2009
DOI:10.1016/j.jmr.2009.07.028
We describe a new, computationally efficient method for computing the molecular alignment tensor based on the molecular shape. The increase in speed is achieved by re-expressing the problem as one of numerical integration, rather than a simple uniform sampling (as in the PALES method), and by using a convex hull rather than a detailed representation of the surface of a molecule. This method is applicable to bicelles, PEG/hexanol, and other alignment media that can be modeled by steric restrictions introduced by a planar barrier. This method is used to further explore and compare various representations of protein shape by an equivalent ellipsoid. We also examine the accuracy of the alignment tensor and residual dipolar couplings (RDC) prediction using various ab initio methods. We separately quantify the inaccuracy in RDC prediction caused by the inaccuracy in the orientation and in the magnitude of the alignment tensor, concluding that orientation accuracy is much more important in accurate prediction of RDCs.
Co-reporter:David Fushman;Yaroslav Ryabov
Magnetic Resonance in Chemistry 2006 Volume 44(Issue S1) pp:S143-S151
Publication Date(Web):6 JUL 2006
DOI:10.1002/mrc.1822
In this paper, we propose the idea that simultaneous analysis of NMR relaxation data and residual dipolar couplings (RDCs) can provide information about interdomain dynamics in a multidomain protein, which cannot be derived from each data set separately. Specifically, such an approach can be useful when the interdomain motions occur on a timescale comparable to or slower than the overall tumbling in solution. We analyze residual dipolar couplings together with 15N relaxation data for Lys48-linked di-ubiquitin (Ub2), in which interdomain dynamics are described as interconversion between two distinct conformational states of the protein. Our results show that 15N relaxation and residual dipolar coupling data can be used as two complementary experimental data sets for consistent characterization of interdomain conformations and dynamics in this dual-domain protein. Copyright © 2006 John Wiley & Sons, Ltd.
Co-reporter:David Fushman, Ranjani Varadan, Michael Assfalg, Olivier Walker
Progress in Nuclear Magnetic Resonance Spectroscopy 2004 Volume 44(3–4) pp:189-214
Publication Date(Web):30 July 2004
DOI:10.1016/j.pnmrs.2004.02.001
Co-reporter:Aydin Haririnia, Mariapina D’Onofrio, David Fushman
Journal of Molecular Biology (4 May 2007) Volume 368(Issue 3) pp:753-766
Publication Date(Web):4 May 2007
DOI:10.1016/j.jmb.2007.02.037
Numerous cellular processes are regulated by (poly)ubiquitin-mediated signaling events, which involve a covalent modification of the substrate protein by a single ubiquitin or a chain of ubiquitin molecules linked via a specific lysine. Remarkably, the outcome of polyubiquitination is linkage-dependent. For example, Lys48-linked chains are the principal signal for proteasomal degradation, while Lys63-linked chains act as nonproteolytic signals. Despite significant progress in characterization of various cellular pathways involving ubiquitin, understanding of the structural details of polyubiquitin chain recognition by downstream cellular effectors is missing. Here we use NMR to study the interaction of a ubiquitin-interacting motif (UIM) of the proteasomal subunit S5a with di-ubiquitin, the simplest model for polyubiquitin chain, to gain insights into the mechanism of polyubiquitin recognition by the proteasome. We have mapped the binding interface and characterized the stoichiometry and the process of UIM binding to Lys48- and Lys63-linked di-ubiquitin chains. Our data provide the first direct evidence that UIM binding involves a conformational transition in Lys48-linked di-ubiquitin, which opens the hydrophobic interdomain interface. This allows UIM to enter the interface and bind directly to the same ubiquitin hydrophobic-patch surface as utilized in UIM:monoubiquitin complexes. The results indicate that up to two UIM molecules can bind di-ubiquitin, and the binding interface between UIM and ubiquitin units in di-ubiquitin is essentially the same for both Lys48- and Lys63-linked chains. Our data suggest possible structural models for the binding of UIM and of full-length S5a to di-ubiquitin.
Co-reporter:David Fushman
Structure (3 January 2017) Volume 25(Issue 1) pp:1-3
Publication Date(Web):3 January 2017
DOI:10.1016/j.str.2016.12.010
Linear (head-to-tail linked) polyubiquitin chains play a key role in the regulation of NF-κB signaling. In this issue of Structure, Lin et al. (2017) shed light on how linear tri-ubiquitin binds to ABIN2, a molecule that shares ubiquitin-binding properties of NEMO, the key activator of NF-κB.
Co-reporter:Carlos A. Castañeda, Tanuja R. Kashyap, Mark A. Nakasone, Susan Krueger, David Fushman
Structure (2 July 2013) Volume 21(Issue 7) pp:1168-1181
Publication Date(Web):2 July 2013
DOI:10.1016/j.str.2013.04.029
•K11-linked Ub2 adopts unique conformations distinct from both K48- and K63-linked Ub2•Solution and crystal structures reveal broad range of K11-linked chain conformations•Presence of salt increases interactions between Ub units and compacts K11-linked Ub2•K11-linked polyUb binds K48- and K63-selective receptors with intermediate affinityK11-linked polyubiquitin chains play important signaling and regulatory roles in both degradative and nonproteolytic pathways in eukaryotes. To understand the structural basis of how these chains are recognized and distinguished from other polyubiquitins, we determined solution structures of K11-linked diubiquitin (K11-Ub2) in the absence and presence of salt. These structures reveal that K11-Ub2 adopts conformations distinct from those of K48-linked or K63-linked chains. Importantly, our solution NMR and SANS data are inconsistent with published crystal structures of K11-Ub2. We found that increasing salt concentration compacts K11-Ub2 and strengthens interactions between the two Ub units. Binding studies indicate that K11-Ub2 interacts with ubiquitin-receptor proteins from both proteasomal and nonproteasomal pathways but with intermediate affinity and different binding modes than either K48-linked or K63-linked diubiquitin. Our data support the hypothesis that polyubiquitin chains of different linkages possess unique conformational and dynamical properties, allowing them to be recognized differently by downstream receptor proteins.Download high-res image (282KB)Download full-size image
Co-reporter:Daoning Zhang, Shahri Raasi, David Fushman
Journal of Molecular Biology (14 March 2008) Volume 377(Issue 1) pp:162-180
Publication Date(Web):14 March 2008
DOI:10.1016/j.jmb.2007.12.029
Ubiquilin/PLIC proteins belong to the family of UBL–UBA proteins implicated in the regulation of the ubiquitin-dependent proteasomal degradation of cellular proteins. A human presenilin-interacting protein, ubiquilin-1, has been suggested as potential therapeutic target for treating Huntington's disease. Ubiquilin's interactions with mono- and polyubiquitins are mediated by its UBA domain, which is one of the tightest ubiquitin binders among known ubiquitin-binding domains. Here we report the three-dimensional structure of the UBA domain of ubiquilin-1 (UQ1-UBA) free in solution and in complex with ubiquitin. UQ1-UBA forms a compact three-helix bundle structurally similar to other known UBAs, and binds to the hydrophobic patch on ubiquitin with a Kd of 20 μM. To gain structural insights into UQ1-UBA's interactions with polyubiquitin chains, we have mapped the binding interface between UQ1-UBA and Lys48- and Lys63-linked di-ubiquitins and characterized the strength of UQ1-UBA binding to these chains. Our NMR data show that UQ1-UBA interacts with the individual ubiquitin units in both chains in a mode similar to its interaction with mono-ubiquitin, although with an improved binding affinity for the chains. Our results indicate that, in contrast to UBA2 of hHR23A that has strong binding preference for Lys48-linked chains, UQ1-UBA shows little or no binding selectivity toward a particular chain linkage or between the two ubiquitin moieties in the same chain. The structural data obtained in this study provide insights into the possible structural reasons for the diversity of polyubiquitin chain recognition by UBA domains.
Co-reporter:Carlos A. Castañeda, Emma K. Dixon, Olivier Walker, Apurva Chaturvedi, ... David Fushman
Structure (1 March 2016) Volume 24(Issue 3) pp:423-436
Publication Date(Web):1 March 2016
DOI:10.1016/j.str.2016.01.007
•Di-Ubs of all possible lysine linkages were made and studied by NMR and DUB assays•K27-Ub2 has unique NMR characteristics, dynamics, and resistance to DUB cleavage•K27-Ub2 adopts open conformations in solution capable of bidentate binding to receptors•K27-Ub2 binds UBA2 domain of hHR23A through bidentate interactions similar to K48-Ub2Polyubiquitination, a critical protein post-translational modification, signals for a diverse set of cellular events via the different isopeptide linkages formed between the C terminus of one ubiquitin (Ub) and the ɛ-amine of K6, K11, K27, K29, K33, K48, or K63 of a second Ub. We assembled di-ubiquitins (Ub2) comprising every lysine linkage and examined them biochemically and structurally. Of these, K27-Ub2 is unique as it is not cleaved by most deubiquitinases. As this remains the only structurally uncharacterized lysine linkage, we comprehensively examined the structures and dynamics of K27-Ub2 using nuclear magnetic resonance, small-angle neutron scattering, and in silico ensemble modeling. Our structural data provide insights into the functional properties of K27-Ub2, in particular that K27-Ub2 may be specifically recognized by K48-selective receptor UBA2 domain from proteasomal shuttle protein hHR23a. Binding studies and mutagenesis confirmed this prediction, further highlighting structural/recognition versatility of polyubiquitins and the potential power of determining function from elucidation of conformational ensembles.
Co-reporter:Carlos A. Castañeda, Apurva Chaturvedi, Christina M. Camara, Joseph E. Curtis, Susan Krueger and David Fushman
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 8) pp:NaN5788-5788
Publication Date(Web):2015/09/21
DOI:10.1039/C5CP04601G
Polyubiquitination is a critical protein post-translational modification involved in a variety of processes in eukaryotic cells. The molecular basis for selective recognition of the polyubiquitin signals by cellular receptors is determined by the conformations polyubiquitin chains adopt; this has been demonstrated for K48- and K63-linked chains. Recent studies of the so-called non-canonical chains (linked via K6, K11, K27, K29, or K33) suggest they play important regulatory roles in growth, development, and immune system pathways, but biophysical studies are needed to elucidate the physical/structural basis of their interactions with receptors. A first step towards this goal is characterization of the conformations these chains adopt in solution. We assembled diubiquitins (Ub2) comprised of every lysine linkage. Using solution NMR measurements, small-angle neutron scattering (SANS), and in silico ensemble generation, we determined population-weighted conformational ensembles that shed light on the structure and dynamics of the non-canonical polyubiquitin chains. We found that polyubiquitin is conformationally heterogeneous, and each chain type exhibits unique conformational ensembles. For example, K6-Ub2 and K11-Ub2 (at physiological salt concentration) are in dynamic equilibrium between at least two conformers, where one exhibits a unique Ub/Ub interface, distinct from that observed in K48-Ub2 but similar to crystal structures of these chains. Conformers for K29-Ub2 and K33-Ub2 resemble recent crystal structures in the ligand-bound state. Remarkably, a number of diubiquitins adopt conformers similar to K48-Ub2 or K63-Ub2, suggesting potential overlap of biological function among different lysine linkages. These studies highlight the potential power of determining function from elucidation of conformational states.

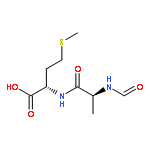
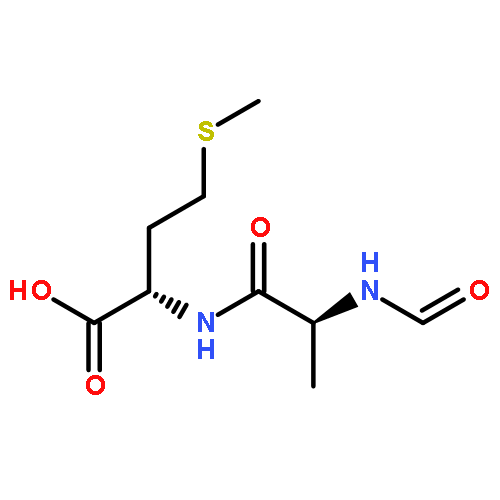
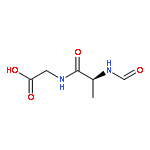
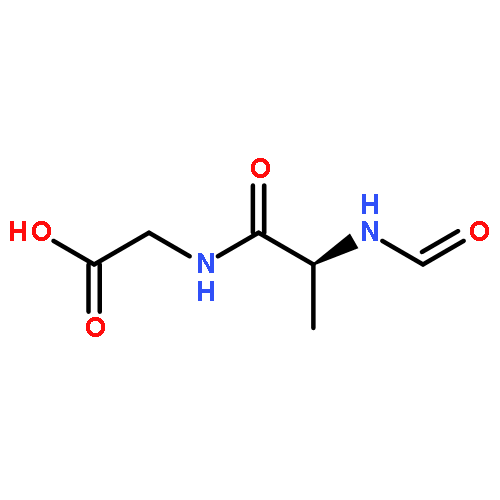


![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6_b.png)