Co-reporter:Riguang Zhang, Guangxiang Wen, Hertanto Adidharma, Armistead G. Russell, Baojun Wang, Maciej Radosz, and Maohong Fan
ACS Catalysis December 1, 2017 Volume 7(Issue 12) pp:8285-8285
Publication Date(Web):October 27, 2017
DOI:10.1021/acscatal.7b02800
Density functional theory (DFT) analysis is used to shed light on the intricate effects of the Co2C and Co/Co2C catalyst crystal facets on the selectivity of the C2 oxygenate and hydrocarbon formation in Fischer–Tropsch synthesis. Three representative low-index Co2C (101), (110), and (111) surfaces, varying in surface energy from low and medium to high, are model examples of different Co2C exposed crystal facets. Since CHx (x = 1–3), CO, and H species are the key intermediates critical to the C2 oxygenate selectivity, all Fischer–Tropsch reactions related to CHx (x = 1–3) species, including CO insertion into CHx (x = 1–3) and CHx + CHy (x, y = 1–3) coupling to form C2 species (C2Hx and C2HxO), as well as the hydrogenation and dissociation of CHx (x = 1–3) to form C1 species (CH4 and C), are used as examples examined at a typical FTS temperature of 493 K. On Co2C (101) and (110) surfaces, CH and CH2 species are dominant form of the CHx species, CH self-coupling to C2H2 and CH coupling with CH2 to CH2CH is dominant C2 species. However, on a Co2C (111) surface, only CH monomer is a dominant CHx (x = 1–3) species, and CO insertion into CH to form CHCO is a dominant C2 species. CH4 and C production on these three surfaces is impossible. These results show that C2 species formation, rather than C1 species, is a preferable pathway on different Co2C crystal facets in FTS reactions. Moreover, the C2 selectivity, quantitatively estimated from the effective barrier difference, is found to be sensitive to the Co2C crystal facet. The Co/Co2C (111) interface catalyst is more favorable for C2 oxygenate formation in comparison to the pure Co2C (111) catalyst, whereas the Co/Co2C (110) and Co/Co2C (101) interface catalysts are unfavorable for C2 oxygenate formation in comparison to the pure Co2C (110) and (101) catalysts. Therefore, for the FTS over Co2C and Co/Co2C catalysts, the Co2C (111) crystal facet is found to have an unexpectedly high selectivity for C2 oxygenates, whereas the Co2C (101) and (110) crystal facets are found to have a high selectivity toward C2 hydrocarbons. The results mean that controlling the crystal facets of Co2C catalysts using well-defined preparation methods can be an effective tool to tune the FTS selectivity toward the most desirable products.Keywords: Co2C; crystal facet; density functional theory; Fischer−Tropsch synthesis; selectivity;
Co-reporter:Yongwu Lu, Riguang Zhang, Baobao Cao, Binghui Ge, Franklin Feng Tao, Junjun Shan, Luan Nguyen, Zhenghong Bao, Tianpin Wu, Jonathan W. Pote, Baojun Wang, and Fei Yu
ACS Catalysis August 4, 2017 Volume 7(Issue 8) pp:5500-5500
Publication Date(Web):June 21, 2017
DOI:10.1021/acscatal.7b01469
CO hydrogenation to higher alcohols (C2+OH) provides a promising route to convert coal, natural gas, shale gas, and biomass feedstocks into value-added chemicals and transportation fuels. However, the development of nonprecious metal catalysts with satisfactory activity and well-defined selectivity toward C2+OH remains challenging and impedes the commercialization of this process. Here, we show that the synergistic geometric and electronic interactions dictate the activity of Cu0–χ-Fe5C2 binary catalysts for selective CO hydrogenation to C2+OH, outperforming silica-supported precious Rh-based catalysts, by using a combination of experimental evidence from bulk, surface-sensitive, and imaging techniques collected on real and high-performance Cu–Fe binary catalytic systems coupled with density functional theory calculations. The closer is the d-band center to the Fermi level of Cu0–χ-Fe5C2(510) surface than those of χ-Fe5C2(510) and Rh(111) surface, and the electron-rich interface of Cu0–χ-Fe5C2(510) due to the delocalized electron transfer from Cu0 atoms, facilitates CO activation and CO insertion into alkyl species to C2-oxygenates at the interface of Cu0–χ-Fe5C2(510) and thus enhances C2H5OH selectivity. Starting from the CHCO intermediate, the proposed reaction pathway for CO hydrogenation to C2H5OH on Cu0–χ-Fe5C2(510) is CHCO + (H) → CH2CO + (H) → CH3CO + (H) → CH3CHO + (H) → CH3CH2O + (H) → C2H5OH. This study may guide the rational design of high-performance binary catalysts made from earth-abundant metals with synergistic interactions for tuning selectivity.Keywords: CO hydrogenation; copper; higher alcohols; Hägg iron carbide; reaction mechanism; synergistic effect;
Co-reporter:Lixia Ling, Zhongbei Zhao, Xue Feng, Qiang Wang, Baojun Wang, Riguang Zhang, and Debao Li
The Journal of Physical Chemistry C August 3, 2017 Volume 121(Issue 30) pp:16399-16399
Publication Date(Web):July 11, 2017
DOI:10.1021/acs.jpcc.7b05024
A periodic density functional theory (DFT) calculation has been used to study the NO reduction by H2 on the stepped Pd(211) surface. The main route of N2 generation changes with temperature increasing. The dimer path is main for the formation of N2 at low temperature, in which two NO react and generate N2O2, and then N2O2 decompose to produce N2. However, the active N path becomes main to generate N2 via NO hydrogenate and dissociate to produce active N at high temperature. The formation of NH3 is via the successive hydrogenation of N or NH. Additionally, energy barriers showed that the Pd(211) surface exhibited higher catalytic activity to the reduction of NO by H2 than that on the Pd(111) surface, and the kinetics showed that the selectivity of N2 is higher than that of NH3 on the stepped Pd(211) surface below about 500 K.
•The surface structure and composition of Ni catalyst alter CH4 formation pathway.•Zr promoter effectively improve the activity and selectivity of CH4 due to no CH3OH yield.•Surface carbon is suppressed on ZrNi(211) due to the higher energy of CH2 dehydrogenation.•The activity and selectivity of CH4 formation follow the order: ZrNi(211) > Ni(211) > Ni(111).•Carbon deposition at the Ni step could be alleviated by hydrogenation under high H2/CO ratio.In order to reveal the underlying mechanism of coke-deposition resistance of the promoter Zr, the comparative studies about the reaction pathways of CO methanation have been carried out over the stepped Ni(211) and Zr-modified Ni(211) surfaces using the density functional theory. DFT results show that CO → COH → C → CH → CH2 → CH3 → CH4 and CO → COH → HCOH → CH2OH → CH2 → CH3 → CH4 are mainly responsible for CH4 formation on Ni(211). Thus, in the former pathway, the main contributor to carbon deposition is the dissociation of COH; while in the latter pathway, carbon formation ascribes to the decomposition of CH2 due to the competition of CH2 between hydrogenation and dehydrogenation. Conclusively, Ni(211) is much susceptible to carbon formation. However, ZrNi(211) surface exhibits high resistance to carbons with the energetically favorable pathway of CO → HCO → CH2O → CH2OH → CH2 → CH3 → CH4 since CH2 prefers to be hydrogenated to CH3 rather than being dehydrogenated into CH. On the other hand, for the effects of surface structure and composition on the selectivity, Ni(211) displays a remarkable increase in selectivity to CH4 compared to Ni(111), which ascribes to much difference in the activity of CH2OH between the dissociation to CH2 and the hydrogenation to CH3OH, the former is superior to the latter. On ZrNi(211), even no CH3OH yield is expected. Further, the presence of promoter Zr greatly decreases the overall activation energy of CH4 formation on ZrNi(211) surface, this results in a significant increase of the reaction activity compared to Ni(111) and Ni(211). To sum up, ZrNi(211) is highly activity and selectivity of CH4 formation in CO methanation, and particularly resistant to carbon formation, which will provide a way of fabricating a more effective Ni-based catalyst.Download full-size image
Chemical Engineering Journal 2017 Volume 308(Volume 308) pp:
Publication Date(Web):15 January 2017
DOI:10.1016/j.cej.2016.09.004
•AC can enhance the stability of magic clusters Pdn (n = 4, 6, 8, 13, 19, 23).•Pdn cluster size influences Hg0 adsorption greatly, and Pd13/AC is optimal.•CuPd12, Cu2Pd11 and Cu6Pd7 bimetallic clusters are relatively easy to obtain.•The adsorption of Hg0 is affected by Cu doping ratio, and Cu2Pd11/AC is the best.A density functional theory (DFT) method was used to study the adsorption of Hg0 on activated carbon supported Pdn (n ⩽ 38) clusters, the size and Cu modulation of Pd clusters were investigated. The adsorption energies of Hg0 on Pdn/AC (n = 4, 6, 8, 13, 19, 23) show that the strongest adsorption strength is on the Pd13/AC. AC can prevent Pd clustering to a bulk and disperse cluster on the surface by comparing the interaction energy of Pdn cluster on AC with the binding energy per atom of corresponding Pdn cluster. Furthermore, some of Pd atoms in Pd13 cluster were substituted by Cu atoms to improve the adsorption of Hg0 and reduce the dosage of Pd. CuPd12, Cu2Pd11 and Cu6Pd7 are the most stable bimetallic clusters. In addition, the adsorption of Hg0 shows that Cu2Pd11/AC is the most effective adsorbent for Hg0.Download high-res image (111KB)Download full-size image
•Fe addition makes metal-support interaction increase in perfect Ni/MgO.•Fe addition makes metal-support interaction reduce in defect Ni/MgO.•H2 dissociation has lower reaction barrier on perfect NiFe/MgO than on defect one.•Medium metal-support interaction can improve catalyst performance.The effect of addition of a second metal to activation component and the support with different morphology on H2 dissociation on Ni/MgO catalyst was investigated using density functional theory. The results show that after addition of a second metal Fe, the metal-support interaction is increased in perfect MgO supported Ni catalyst while it is decreased in defective MgO supported Ni catalyst. Although the change is difference between metal-support interaction, the ability of H2 dissociation is increased on the two surfaces. Analyzing those results from addition of Fe, Co and Cu to Ni/MgO, one can conclude that it is neither too strong nor too weak (i.e., medium) interaction between metal and support that favors to improve the catalyst performance.Download high-res image (93KB)Download full-size image
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 35) pp:24357-24368
Publication Date(Web):2017/09/13
DOI:10.1039/C7CP02579C
The adsorption and reactions of CO2 and H2O on both monoclinic and hexagonal crystal K2CO3 were investigated using the density functional theory (DFT) approach. The calculated adsorption energies showed that adsorption of H2O molecules was clearly substantially stronger on the K2CO3 surface than the adsorption of CO2, except on the (001)-1 surface of hexagonal K2CO3, where CO2 is competitively adsorbed with H2O. Carbonation reactions easily occur on pure K2CO3 and involve two parallel paths: one is where adsorbed H2O reacts with molecular CO2 in gas to form the bicarbonate, while the other is where H2O dissociates into OH and H before bicarbonate formation, and then OH reacts with gaseous CO2 to form a bicarbonate. Our results indicate that adding a support or promoter or using a special technique to expose more (001)-1 surfaces in hexagonal K2CO3 may improve the conversion of CO2 to the bicarbonate, which provides a theoretical direction for the experimental preparation of the K2CO3 sorbent to capture CO2.
Co-reporter:Lixia Ling;Qiang Wang;Riguang Zhang;Debao Li
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 45) pp:30883-30894
Publication Date(Web):2017/11/22
DOI:10.1039/C7CP05411D
In this study, the formation mechanism of C2 oxygenates and ethanol from syngas on Fe-decorated Cu bimetallic catalyst was investigated using density functional theory (DFT) calculations together with microkinetic modeling. The results showed that CH2 was the most favored monomer among all the CHx (x = 1–3) species over the FeCu bimetallic catalyst, which was more favorable than CH3OH formation. Namely, the FeCu catalyst exhibited a good selectivity toward CH2 formation instead of CH3OH formation in syngas conversion. Starting from the CH2 monomer, CH2CO and CH3CO via CO insertion into CH2 and CH2CO hydrogenation were the major products instead of C2 hydrocarbons or methane, CH3CO was successively hydrogenated to ethanol via CH3CHO and CH3CH2O intermediates. Moreover, the microkinetic modeling showed that the FeCu bimetallic catalyst had a high selectivity toward ethanol rather than methanol and methane. Further, the addition of Fe into the Cu catalyst promoted CHx formation by accelerating C–O bond cleavage, suppressed methanol formation, and facilitated C2 oxygenate formation rather than methane formation, suggesting that the synergetic effect between Fe and Cu played an important role in the formation of C2 oxygenates and ethanol. In addition, it is believed that the insights derived from this study can provide clues for the catalyst design of oxygenate synthesis and other bimetallic catalytic systems.
The catalytic selectivity, the functions of a TiO2 support and promoter, and the mechanism of ethanol synthesis from syngas on a Rh/TiO2 model catalyst have been fully identified. Our results show that all species preferentially interact with Rh7 clusters of a Rh/TiO2 catalyst, rather than the support and cluster–support interface. CO → CHO → CH2O → CH3O is an optimal pathway. CH3 formed via the CH3O → CH3 + O route is the most favored CHx (x = 1–3) monomer, and this route is more favorable than methanol formation by CH3O hydrogenation; CO insertion into CH3 can then form CH3CO, followed by successive hydrogenation to ethanol. Methane is formed by CH3 hydrogenation. The Rh/TiO2 catalyst exhibits better catalytic activity and selectivity toward CH3 than CH3OH formation. Starting from the CH3 species, CH4 formation is more favorable than CH3CO formation; thus, ethanol productivity and selectivity on a Rh/TiO2 catalyst with a support is determined only by CH4 formation, which is similar to that on a pure Rh catalyst without a support. Introducing an Fe promoter into the Rh/TiO2 catalyst effectively suppresses methane production, and promotes CH3CO formation. Therefore, compared to a pure Rh catalyst without a support, the TiO2 support serves only to promote the activity and selectivity of CH3 formation, and provide more CH3 species for ethanol formation; methane formation is independent of the Rh catalyst support, and depends only on the promoter. In order to achieve high ethanol productivity and selectivity, an effective Rh-based catalyst must contain a suitable combination of supports and promoters, in which the promoter, M, should have characteristics that draw the d-band center of the MRh/TiO2 catalyst closer to the Fermi level compared to the Rh7/TiO2 catalyst; as a result, the MRh/TiO2 catalyst can suppress CH4 production and facilitate C2 oxygenate formation.
DFT calculations, together with microkinetic modeling, have been employed to probe into the preferred mechanism of hydrocarbon C–C chain growth on Co(10−11) surfaces during Fischer–Tropsch synthesis. The results show that both CH and CH2 are favored CHx (x = 1–3) monomers, and are much easier to be formed than CH4 and CH3OH. CH and CH2 self-coupling via a carbide mechanism realizes the initial C–C chain formation, rather than via a CO/CHO insertion mechanism. Meanwhile, CH3CH2 is the favored C2 monomer, and is predominantly formed via a carbide mechanism rather than via a CO/CHO insertion mechanism, leading to C2H5OH formation. Starting from CH3CH2 intermediates, CH3CH2 coupling with CH2 to form CH3CH2CH2 realizes further C–C chain growth from C2 to C3 species, instead of a CO/CHO insertion mechanism leading to C3H7OH formation. Thus, the proposed mechanism of C–C chain growth is that RCH2CH2 coupling with CH2 to R′CH2CH2 (R′ = RCH2) realizes C–C chain growth. Meanwhile, CHO insertion into RCH2CH leads to RCH2CHCHO, followed by its hydrogenation to an alcohol. However, microkinetic modeling shows that the effect of CH4 formation on the production of C2+ hydrocarbons should be considered, whereas alcohols have a negligible effect on the selectivity of C2+ hydrocarbons. Our results confirm that Co(10−11) surfaces exhibit a better catalytic activity and selectivity toward C2+ hydrocarbon formation.
•Perfect NiCu-MgO interaction is weaker than that in defective one.•Interaction of H and P(Ni2Cu2) is weaker than that of H and D(Ni2Cu2).•H2 dissociation is not easier on P(Ni2Cu2) than that on D(Ni2Cu2).•Addition of Cu to perfect Ni/MgO has no effect on H2 dissociation.•Addition of Cu to D(Ni4) causes H2 dissociation to difficult.Density-functional theory has been conducted to investigate the interactions of bimetal NiCu with MgO at the atomic and electronic level as well as its effects of H adsorption and H2 dissociation, compared with those on Ni/MgO. Two models including Ni2Cu2 cluster supported on perfect MgO(001) and oxygen-vacancy MgO(001) are built to represent bimetal NiCu anchored on MgO catalysts. The results show that there is stronger metal-support interaction in Ni2Cu2/MgO catalyst with oxygen-vacancy than that in perfect Ni2Cu2/MgO catalyst, and the H adsorption on defective Ni2Cu2/MgO is stronger than that on perfect Ni2Cu2/MgO. Correspondingly, H2 dissociation is faster on defective Ni2Cu2/MgO than that on perfect Ni2Cu2/MgO. Compared with Ni4/MgO, the interaction between metal and support is weaker on the corresponding Ni2Cu2/MgO, and the interaction of H and substrate is stronger, moreover, the H2 dissociation is faster on defective Ni2Cu2/MgO while it is not changed on perfect Ni2Cu2/MgO. The results indicate that the addition of Cu to defective Ni/MgO can improve the dissociation of H2, which provides a clue for tuning catalyst performance by modifying the metal-support interaction.Download full-size image
Co-reporter:Lixia Ling, Zhongbei Zhao, Baojun Wang, Maohong Fan and Riguang Zhang
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 16) pp:11150-11156
Publication Date(Web):22 Mar 2016
DOI:10.1039/C6CP01422D
The density functional theory (DFT) method has been performed to study the effects of CO and CO2 on the desulfurization of H2S over a ZnO sorbent. It shows that COS is inevitably formed on the ZnO(100) surface, which tends to be adsorbed onto the surface via a S–C bond binding with either a long or a short Zn–O bond. Potential energy profiles for the COS formation via reactions between H2S and CO, and H2S and CO2 on the ZnO(100) surface have been constructed. In the presence of CO, the dissociated active S of H2S reacting with CO leads to the formation of COS, and the activation energy of the rate-determining step is 87.7 kJ mol−1. When CO2 is present, the linear CO2 is first transferred to active CO2 in a triplet state, and then combines with active S to form COS with the highest energy barrier of 142.4 kJ mol−1. Rate constants at different temperatures show that the formation of COS via the reaction of CO and H2S is easier than that of CO2 and H2S over the ZnO surface.
Co-reporter:Xiaobin Hao, Baojun Wang, Qiang Wang, Riguang Zhang and Debao Li
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 26) pp:17606-17618
Publication Date(Web):20 May 2016
DOI:10.1039/C6CP01689H
CO adsorption and activation on Ni(100), (110) and (111) surfaces have been systematically investigated to probe the effect of coverage and surface structure on CO adsorption and activation. Herein, dispersion-corrected density functional theory calculations (DFT-D) were employed, and the related thermodynamic energies at 523 K were calculated by including the zero-point energy, thermal energy and entropic corrections; the results show that the saturated coverage of CO on the Ni(111), (100) and (110) surfaces correspond to 8/9, 9/12 and 9/9 ML, respectively. As the coverage increases, the stepwise adsorption free energies decrease on the flat (111) and (100) surfaces, whereas small changes occur on the corrugated (110) surface. CO migrates from the three-fold hollow site to the top site on the (111) surface, and from the four-fold hollow to the two-fold bridge site on the (100) surface, while all the CO molecules remain at the short-bridge site on the (110) surface. As a result, the obtained intermolecular CO–CO repulsive interactions on the flat surface are stronger than the interactions on the corrugated surface. Furthermore, the computed CO vibrational frequencies at different levels of coverage over the Ni surfaces agree well with the experimental results. On the other hand, kinetic analyses were utilized to compare the stepwise CO desorption with the dissociation at different degrees of coverage on the three Ni surfaces. CO desorption is more favorable than its dissociation at all coverage levels on the most exposed Ni(111) surface. Analogously, CO desorption becomes more favorable than its dissociation on the Ni(110) surface at higher coverage, except for coverage of 1/9 ML, in which CO desorption competes with its dissociation. However, on the Ni(100) surface, CO dissociation is more favorable than its desorption at 1/12 ML; when the coverage increases from 2/12 to 3/12 ML, equilibrium states exist between dissociation and desorption over the surface; when the coverage is greater than or equal to 4/9 ML, CO desorption becomes more favorable than dissociation. By applying the atomistic thermodynamics method, the determination of stable coverage as a function of temperature and partial pressure provides useful information, not only for surface science studies under ultrahigh vacuum conditions, but also for practical applications at high temperature and pressure in exploring reactions.
The Journal of Physical Chemistry C 2016 Volume 120(Issue 4) pp:2234-2246
Publication Date(Web):January 19, 2016
DOI:10.1021/acs.jpcc.5b10831
Density functional theory calculations have been employed to probe into a comparative study of HCOOH oxidation on Pt(111) and three PtAu(111) surfaces with different Pt atomic ensembles-decorated Au(111) surfaces, denoted as Pt ML, Pt6Au3, and Pt3Au6, respectively. Our results show that HCOOH dehydrogenation is dominant on PtAu(111) surface; as compared to Pt(111) surface, PtAu(111) surfaces are efficient for HCOOH dehydrogenation to CO2, and show an inhibiting effect to HCOOH dehydration to CO. The catalytic activity and selectivity of PtAu(111) surfaces toward HCOOH oxidation are obviously dependent on the Pt atomic ensemble of the reaction active center. The enhanced catalytic activity and selectivity of PtAu bimetallic catalysts toward HCOOH oxidation should be attributed to their suppression to the dehydration reactions of HCOOH, which is well confirmed by the reported experimental results that the high catalytic activity of PtAu catalyst is caused by the increased selectivity toward HCOOH dehydrogenation. It is found that Pt ML surface, consisting of 1.0 monolayer Pt on an Au substrate, is not only efficient in the utilization of Pt, but also highly active and selective toward the dehydrogenation of HCOOH oxidation, which can suppress the formation of poisonous species CO. In addition, the present results may be helpful for the search of prospective substitutes to Pt electrode for hydrogen production in the direct formic acid fuel cell.
Co-reporter:Lixia Ling, Maohong Fan, Baojun Wang and Riguang Zhang
Energy & Environmental Science 2015 vol. 8(Issue 11) pp:3109-3133
Publication Date(Web):04 Aug 2015
DOI:10.1039/C5EE02255J
The control of mercury in flue gas is challenging, and many investigators have focused on different mercury removal technologies. The application of computational chemistry in understanding mercury removal mechanisms will help to modify and design mercury removal materials, thereby improving the efficiency of the removal of mercury in flue gas. Therefore, a review of theoretical studies on the adsorption and oxidation of mercury has been undertaken in the current study. In this contribution, the homogeneous oxidation mechanisms of Hg0 as well as heterogeneous interactions including adsorption of mercury species and oxidation of Hg0 on activated carbon, metals, metal oxides and other materials have been summarized. In addition, possible future directions of theoretical calculations on understanding the removal of mercury are outlined.
Co-reporter:Xiaoqiang Guo, Hongyan Liu, Baojun Wang, Qiang Wang and Riguang Zhang
RSC Advances 2015 vol. 5(Issue 26) pp:19970-19982
Publication Date(Web):05 Feb 2015
DOI:10.1039/C4RA15555F
A density-functional theory (DFT) method has been performed to investigate the reaction of C + O(OH) on three types of bimetallic alloy CoNi(111) surface, and the results obtained are compared with those on the pure Ni(111) surface. Our results show that the introduction of Co into the Ni catalyst is beneficial for the adsorption of C, O and OH species, while it weakens the adsorption of CO. Moreover, O(OH) absorbs preferentially on the CoNi(111) surfaces with the surface enrichment of Co compared with the homogeneous CoNi(111) surface; the increased degree of O adsorption energy outweighs the corresponding values of C on the pure Ni(111) and three types of bimetallic alloy CoNi(111) surfaces, indicating that Co has a stronger affinity for oxygen species than for carbon species. On the other hand, the mechanism of the C + O(OH) reaction and the corresponding rate constants at different temperatures show that OH species have a stronger ability to eliminate carbon than O species on Ni(111) and CoNi(111) surfaces; on the CoNi(111) surface, when the Co surface coverage is equal to 1 monolayer (ML), compared to Ni(111), the C + O reaction can be accelerated. When the Co surface coverage is equal to 3/4 ML, the C + OH reaction is the most favorable; further, the rate constant for the C + OH reaction on a CoNi(111) with Co surface coverage of 3/4 ML is much larger than that of the C + O reaction on a CoNi(111) with Co surface coverage of 1 ML. As a result, for carbon elimination on the CoNi alloy surface, OH species should serve as the key species for carbon elimination, and the Co surface coverage of CoNi(111) surface should be kept at 3/4 ML.
Co-reporter:Cuimei Zhi, Qiang Wang, Baojun Wang, Debao Li and Riguang Zhang
RSC Advances 2015 vol. 5(Issue 82) pp:66742-66756
Publication Date(Web):29 Jul 2015
DOI:10.1039/C4RA17096B
The mechanism of methane synthesis from syngas on a Ni(111) surface has been systematically investigated using the density functional theory method together with the periodic slab model, which covers the main existence form of CHx (x = 1–3) species and all possible formation pathways. Our results show that CO hydrogenation to a HCO intermediate is more favorable than CO desorption and CO direct dissociation on Ni(111); starting from HCO, six possible formation pathways of CHx (x = 1–3) species are considered: (i) HCO → CH, (ii) HCO → HCOH → CH, (iii) HCO → HCOH → CH2OH → CH2, (iv) HCO → CH2O → CH2, (v) HCO → CH2O → CH2OH → CH2, (vi) HCO → CH2O → CH3O → CH3, followed by CHx (x = 1–3) successive hydrogenation to CH4, suggesting that CH is the dominate existing form of CHx (x = 1–3) species involving in CH4 formation from syngas, which is dominantly formed by two parallel reaction pathways (i) and (ii), followed by the sequential hydrogenation of CH species to CH4. Meanwhile, surface C is mainly formed by the pathway of CO → HCO (HCOH) → CH → C. Furthermore, beginning with the key CH intermediate, CH preferred to be hydrogenated to CH2 rather than being dissociated into C.
The Journal of Physical Chemistry C 2015 Volume 119(Issue 19) pp:10355-10364
Publication Date(Web):April 24, 2015
DOI:10.1021/jp512504b
The adsorption and dissociation of H2 with different coverages over the Rh(100) surface have been systematically investigated to probe into the effect of coverage on H2 adsorption and dissociation. Here, the results are obtained using the density functional theory (DFT) method together with the periodic slab model. Both the parallel and vertical modes of H2 adsorption on the Rh(100) surface have been identified, and the detailed studies corresponding to H2 adsorption and dissociation at different coverages are presented. Our results show that the parallel mode of a single H2 adsorbed on the Rh(100) surface is more favorable than the vertical mode, in which the top site is the most stable adsorption site. However, the parallel modes of single H2 adsorbed at the bridge and 4F hollow sites, as well as the vertical mode of single H2 adsorbed at the 4F hollow site are all the dissociative adsorption. On the other hand, with the increasing of H2 coverage from low to high, the most stable adsorption configurations of H2 is the parallel adsorption mode at the top site, and the adsorption energies of these adsorbed H2 molecules will decrease gradually until the saturated adsorption with H2 coverage of 6/12 ML, further, the dissociation of these adsorbed H2 molecules is more favorable both kinetically and thermodynamically than their desorption, suggesting that the dissociation of the adsorbed H2 molecule is more favored than their desorption. Finally, considering the dissociative adsorption of the single H2 molecule with the parallel modes at the bridge and 4F hollow sites, as well as the vertical mode at the 4F hollow site, our results still show that the adsorptions of H2 with different coverages at these sites are still the dissociative adsorption with the dissociative H atoms adsorbed on the Rh surface. Therefore, H2 dominantly exists in the form of H atoms on Rh catalyst under realistic conditions.
The Journal of Physical Chemistry C 2015 Volume 119(Issue 25) pp:14135-14144
Publication Date(Web):June 2, 2015
DOI:10.1021/acs.jpcc.5b03868
A density functional theory (DFT) calculation has been carried out to systematically investigate the mechanism of surface carbon elimination by O and OH on both the alloy FeNi(111) and CuNi(111) surfaces, including the homogeneous and the segregated surfaces, respectively; meanwhile, the obtained results are compared with those on the pure Ni(111) surface in order to probe into the effects of CuNi(111) and FeNi(111) surface structure and second metal composition on the performance of surface carbon elimination. Our results show that compared to the pure Ni(111) surface, the introduction of Fe into Ni increases the adsorption of O, OH, and C species, while it weakens the adsorption of CO and COH; the incorporation of Cu into Ni decreases the adsorption ability of C, O, OH, CO, and COH species. The mechanism of surface carbon elimination by O and OH shows that OH species is more effective for carbon elimination than O species on Ni(111), CuNi(111) surface and the segregated FeNi(111) surface; meanwhile, CuNi(111) and FeNi(111) surface structure and second metal composition have obvious effect on the performance of carbon elimination. Compared to Ni(111), FeNi(111) surface is not favorable for carbon elimination, while CuNi(111) surface is beneficial for carbon elimination, in which the Cu enriched surface is much more favorable than the 1:1 Cu surface and the pure Ni(111), indicating that the segregated CuNi(111) surface with Cu enrichment significantly accelerates carbon elimination. Moreover, the good linear relationship exists between the average adsorption energy of C + O or C + OH and the activation barrier of the C + O(OH) reaction. As a result, once carbon is formed on the segregated CuNi alloy surface with Cu enrichment, carbon deposits can be timely eliminated, which can well explain the reported experimental facts that CuNi bimetallic catalysts with Cu surface enrichment display excellent carbon-resistance ability in CH4/CO2 reforming.
The Journal of Physical Chemistry C 2015 Volume 119(Issue 14) pp:7678-7688
Publication Date(Web):March 19, 2015
DOI:10.1021/jp511289k
The periodic DFT+U calculation has been carried out to elucidate the desulfurization mechanism of H2S with the ceria (110) surface. The calculations show that the H2S molecule dissociatively adsorbs on the stoichiometric and reduced ceria (110) surfaces, respectively. The desulfurization process with the ceria (110) surface mainly contains two steps. First of all, H2S interacts with the stoichiometric surface, and the SO2-forming path is the most favorable reaction route based on the kinetics analysis. Meanwhile, surface O vacancies are created, and the reduced surface is formed. In this process, S in H2S is transferred to another sulfur-containing compound, SO2, which could not reduce the sulfur content in coal gas. Second, S in H2S is captured by filling into the O vacancy and forming CeOS species on the reduced surface. Therefore, it can be inferred that the key to the desulfurization of H2S in coal gas with ceria (110) is that more surface O vacancies are created.
•Theoretical calculations illustrated the mechanisms of enhanced activity for CuY.•The constructed CuY models reflect the various environments around the active center.•Cu+ at the adjacent site II exhibits excellent activity for the active center Cu+.•Introducing Cs effectively enhanced the catalytic activity of CuY zeolites.A density functional theory method has been used to investigate the effect of environment around the active center Cu+ species on the catalytic activity in the oxidative carbonylation of methanol to dimethyl carbonate over CuY zeolites. Based on the configuration of Cu+ located in the supercage, Cu+ or Cs+ species at the sites adjacent to the active center Cu+ species in the supercage are considered as the surrounding environment. The results indicate that the presence of Cu+ in the supercage adjacent to the active center improves the adsorption energy of co-adsorbed CO and elongates the CuOCH3 bond in co-adsorbed CO/CH3O system, stabilizes the transition state for the reaction of CO insertion, and ultimately makes the active center Cu+ species exhibit better catalytic activity. Whereas, Cu+ species at adjacent site in the smallcage plays an opposite role. More importantly, introducing Cs species into the supercage of CuY zeolite significantly improves the adsorption energy of co-adsorbed CO and the stability of transition state configuration for CO insertion reaction, thus, leads to the best catalytic performance among four types of catalysts, which is consistent with the previous experimental results.
•Both sulfurized surfaces have two competitive regeneration pathways.•The rate-determining step for sulfide surfaces is the dissociation of O2.•The regeneration of S-contained surface is easier than S-adsorbed surface.•The O vacancy is favorable to the regeneration of the Fe2O3(0001) surface.The regeneration mechanisms of sulfurized α-Fe2O3 desulfurization sorbent under O2 atmosphere are systematically studied by density functional theory (DFT) slab calculation. The regeneration pathways are proposed for two sulfurized surfaces, “S-contained surface” and “S-adsorbed surface”, which are derived from the substitution of surface O atom by S atom and the adsorption of S atom on the surface Fe atom, respectively. Our results show that two competitive regeneration pathways exist on each sulfurized surface. For the regeneration on the S-contained surface and the S-adsorbed surface, the rate-determining step is the dissociation of O2 with the energy barriers of 136.7 and 227.3 kJ·mol− 1, respectively, suggesting that the regeneration on the S-contained surface is easier than that on the S-adsorbed surface. Then, the O vacancy on the α-Fe2O3(0001) surface can be repaired under O2 atmosphere, and the reparation mechanism demonstrates that the dissociation of O2 on the O-vacancy α-Fe2O3(0001) surface only needs to overcome a lower energy barrier of 49.8 kJ·mol− 1 than those (136.7 and 227.3 kJ·mol− 1) on two sulfurized surfaces. As a result, the presence of O vacancy on the α-Fe2O3(0001) surface can accelerate the dissociation of O2, which is favorable to the regeneration of sulfurized surfaces.
Density functional calculations have been carried out to investigate the source and major species of CHx (x = 1–3) involved in acetic acid synthesis from methane–syngas on the Rh(111) surface. All possible formation pathways of CHx (x = 1–3) from methane and syngas have been systematically investigated. For CHx formation from methane, our results show that CH is the most abundant species; for CHx formation from syngas, all CHx (x = 1–3) species form from CHO by CO hydrogenation, and the optimal formation routes of CHx show that CH and CH3 are the most abundant species rather than CH2 and CH3OH. On the other hand, CH formed by methane is more favourable than CH and CH3 formed by syngas; meanwhile, CO insertion into CHx species to form C2 oxygenates as acetic acid precursors is more favourable than CO hydrogenation to CH and CH3. As a result, in acetic acid synthesis from methane–syngas, CHx (x = 1–3) species come from methane rather than syngas, and the corresponding primary species is CH. In addition, the CO in syngas is predominantly responsible for insertion reactions that produce CHCO, which is a C2 oxygenate precursor leading to the formation of acetic acid. Furthermore, microkinetic modelling analysis shows that the major product of acetic acid synthesis from methane–syngas on the Rh(111) surface is CH3COOH, and that the production of CH3OH cannot compete with that of CH3COOH.
Co-reporter:Lixia Ling, Jiajia Song, Senpeng Zhao, Riguang Zhang and Baojun Wang
RSC Advances 2014 vol. 4(Issue 43) pp:22411-22418
Publication Date(Web):09 May 2014
DOI:10.1039/C4RA02485K
The adsorption and decomposition mechanisms of H2S on different α-Fe2O3(0001) surfaces, including Fe-vacancy, O-vacancy, sulfurized and Cu-, Zn- and Co-doped surfaces, have been studied systematically using periodic density functional calculations. The results show that the Fe-vacancy surface exhibits an excellent catalytic activity towards the decomposition of H2S, which is favorable for the desulfurization. Both O-vacancy and sulfurized surfaces have negative effects on the desulfurization. The doping of Cu, Zn and Co on the α-Fe2O3(0001) surface is beneficial to enhance the desulfurization performance of the hematite sorbent, of which Zn addition is a comparatively good candidate taking desulfurization efficiency and economic factors into account.
Co-reporter:Riguang Zhang ; Guiru Wang ; Baojun Wang ;Lixia Ling
The Journal of Physical Chemistry C 2014 Volume 118(Issue 10) pp:5243-5254
Publication Date(Web):February 21, 2014
DOI:10.1021/jp409447u
Density functional theory calculations have been employed to investigate the effect of promoter Mn on ethanol formation from syngas on a Mn-promoted MnCu(211) surface. Our results show that CO + 3H → CHO + 2H → CH2O + H → CH3O is an optimal pathway for the overall CO conversion. Starting with CH3O, CH3 is formed via CH3O → CH3 + O. Then, CHO insertion into CH3 can form CH3CHO, and further, CH3CHO is successively hydrogenated to ethanol via CH3CH2O intermediate. Meanwhile, CH3OH is formed via CH3O + H → CH3OH. Compared to the pure Cu(211) surface, CH3 formation is found to be energetically compatible with CH3OH formation on the MnCu(211) surface, which can lead to more CH3 sources and less CH3OH; thus, the productivity and selectivity of ethanol can be improved. On the other hand, starting from CH3, the MnCu(211) surface is more favorable for CHO insertion into CH3 to CH3CHO in comparison with CH3 hydrogenation, dissociation and coupling to CH4, CH2, and C2H6 due to their high activation barriers; namely, the MnCu(211) surface exhibits a better selectivity toward C2 oxygenates rather than hydrocarbons. As a result, we can show that, by introducing promoter Mn into Cu catalyst, the productivity and selectivity to ethanol from syngas can be effectively improved.
Co-reporter:Hongyan Liu, Botao Teng, Maohong Fan, Baojun Wang, Yulong Zhang, H. Gordon Harris
Fuel 2014 Volume 123() pp:285-292
Publication Date(Web):1 May 2014
DOI:10.1016/j.fuel.2014.01.087
•DFT method is employed to investigate the interaction between Ni and MgO.•Charge transfers from MgO to adspecies through Ni are studied.•Strong Ni–MgO interaction can increase the dissociation activity of CH4.•Calculation confirms that strong Ni–MgO interaction can resist carbon deposition.•New design approaches for high activity and free-carbon catalysts are proposed.A theoretical understanding of CH4 dissociation on Ni-based catalysts is of great importance for the development of CH4 reforming catalysts with high activity and carbon-deposition resistance. Based on comparisons of CH4 dissociation on perfect and defective MgO supported Ni4, as well as Ni(1 1 1), the effects of the strong interactions between Ni4 and MgO on CH4 dissociation are systematically investigated by density functional theory (DFT) calculations. Our results indicate that the interaction between Ni4 and the defective MgO is stronger than for the perfect MgO. Consequently, the adsorptions of CHx(x = 0–4) are weaker than those on the perfect Ni4/MgO. Hirshfeld charge analysis shows that electrons are transferred from MgO to Ni4, then to CHx adspecies; the stronger interactions between Ni4 and MgO lead to less electronic transfer from Ni4 to adspecies, which result in weaker adsorption of CHx. Potential energy surface calculations of CH4 dissociation indicate that there are lower energy barriers for the sequent dissociations of CH4 → CH2 + 2H and an appropriate barrier of CH oxidation matching up with that of CH2 further dissociation on the model catalyst of Ni4 supported on defective MgO. This might be an elementary requirement for an excellent CH4 reforming catalyst, and may shed light on experimental catalyst development.
The sulfurization mechanism of H2S on the ZnO(101¯0) surface during the desulfurization of coal gas was investigated by using periodic density functional theory (DFT) calculations. The adsorption of H2S, SH, atomic S, and atomic H, as well as the coadsorption of SH and an H atom, and the coadsorption of S and two H atoms, were initially examined to identify energetically favorable intermediates. Potential energy profiles for three paths of H2S − ZnO(101¯0) interactions producing H2 and H2O were obtained, respectively. Our results show that H2S is preferred to dissociatively adsorb on the ZnO(101¯0) surface, followed by dehydrogenation process to form sulfur species. Molecular-level calculations demonstrate that H2O formation via the H2S−ZnO interaction is the most probable reaction pathway both kinetically and thermodynamically. ZnO has double functions during the desulfurization of H2S. One is as a catalyst to accelerate the dissociation of H2S, while the other is as the reactant participating in the reaction of H2S with ZnO to form H2O.Highlights► ZnO has two roles (as catalyst and reactant) during the desulfurization of H2S. ► H2S is preferred to dissociatively adsorb on the ZnO(101(—)0) surface. ► The S atom bridging a Zn−O bond is the most stable configuration. ► H2O-forming via H2S-ZnO interaction is the most favorable reaction route.
•H2S adsorbs on the α-Fe2O3(0001) surface in molecular mode.•The pathways for H2-forming and H2O-forming are competitive kinetically.•α-Fe2O3 plays two roles (as catalyst and reactant) in the interaction with H2S.The interaction mechanism of H2S and the α-Fe2O3(0001) surface during the desulfurization has been investigated by the density functional theory (DFT) method within a periodic slab model. Adsorptions of H2S, SH, S and H on the α-Fe2O3(0001) surface have been initially examined. Our results show that H2S, SH and atomic S favorably adsorb on the top of Fe site, and atomic H lies on the top of O site. Potential energy profiles have been constructed for the interactions of H2S with the α-Fe2O3(0001) surface along with two channels producing H2 and H2O. The calculations show that H2S firstly adsorbs on the α-Fe2O3(0001) surface in molecular mode, followed by two dehydrogenation processes and forming surface sulfur species. Further, the processes of H-migration lead to the formation of H2 or H2O. Molecular-level calculations demonstrate that the pathways of H2-forming and H2O-forming are competitive kinetically. Two roles of the α-Fe2O3 during the interactions between H2S and the α-Fe2O3(0001) surface have also been discussed.
The regeneration mechanisms of the sulfurized and oxygen-deficient ZnO(101¯0) surfaces in an oxygen atmosphere have been systematically studied by using the density functional theory (DFT) method. An activation energy of 36.79 kJ·mol− 1 is needed for the regeneration of the sulfurized ZnO(101¯0) surface at the GGA–PW91 functional level. The formed SO2 lies on the ZnO(101¯0) surface horizontally, S in SO2 bonds to a surface oxygen atom to form an analogical SO3 structure. Two regeneration mechanisms are studied for the oxygen-deficient ZnO(101¯0) surface. One is that O2 dissociatively adsorbs on the oxygen-deficient ZnO(101¯0) surface leading to the regeneration of the surface. The other is that O2 molecularly adsorbs on the oxygen-deficient ZnO(101¯0) surface, then a little activation energy of 29.43 kJ·mol− 1 is needed to make the surface regenerate. It can be concluded that the sulfurized and oxygen-deficient ZnO(101¯0) surfaces are easy to be regenerated in an atmosphere containing O2.Highlights► The sulfurized ZnO(101¯0) surface is easy to be regenerated in the presence of O2. ► SO2 prefers to lie on the ZnO(101¯0) surface horizontally. ► The oxygen-deficient ZnO(101¯0) surface can be regenerated easily.
Co-reporter:Riguang Zhang, Jingrui Li and Baojun Wang
RSC Advances 2013 vol. 3(Issue 30) pp:12287-12298
Publication Date(Web):23 Apr 2013
DOI:10.1039/C3RA40256H
Cu-exchanged Y zeolites with different Si/Al ratios have been investigated using a density functional theory method in order to determine the effect of Si/Al ratios on the catalytic activity for producing DMC by the oxidative carbonylation of methanol. The stable structures and catalytic activity of the CuY zeolites with different Si/Al ratios are identified. In addition, CO adsorption and the effect of CH3O on CO adsorption, including the changes in the adsorption energy and stretching vibrational frequency for the adsorbed CO, are obtained. Our results indicate that the CuY zeolite with a Si/Al ratio = 6.5 has the highest catalytic activity for producing DMC by oxidative carbonylation of methanol among all the Si/Al ratios considered in this work, which is supported by the experimental facts. Finally, our results suggest that theoretical calculations can be used as a useful tool and provide good theoretical guidance for the experimental design of CuY zeolites.
Co-reporter:Hongyan Liu, Baojun Wang, Maohong Fan, Neil Henson, Yulong Zhang, Brian Francis Towler, H. Gordon Harris
Fuel 2013 Volume 113() pp:712-718
Publication Date(Web):November 2013
DOI:10.1016/j.fuel.2013.06.022
•Propose a theoretical basis for catalyst design.•The activation energy of CH4 dissociation is predicted without C deposition.•Using adsorption energy to instead of the properties of catalyst surfaces.Density functional theory (DFT) has been applied to investigate the adsorptions of CH4, CH3, CH2, CH, C, H and dissociations CH4, CH3, CH2, CH on the (1 1 1) catalyst surfaces of elementary metals Co bimetals NiFe. More important, the adsorptions and dissociations of those adspecies on elementary metals (Fe, Co, Ni and Cu) and bimetals (NiFe, NiCo and NiCu) have been analyzed. The adsorption energies, activation energies, reaction energies and d-band centers of the catalysts were calculated and their linear correlations were established. The adsorption energy decreases with the d-band center of the catalyst surface shift away from the Fermi level, and thus the increase in activation energy and reaction energy. Therefore, a good catalyst should have a moderate d-band center in CH4/CO2 reforming. This research finds that segregated NiCu is the best one among the eight CH4/CO2 reforming catalysts, Fe, Co, Ni, Cu, NiFe, NiCo, NiCu, and NiCu(S) [segregated NiCu].Graphical abstract
Co-reporter:Riguang Zhang, Xuancheng Sun, and Baojun Wang
The Journal of Physical Chemistry C 2013 Volume 117(Issue 13) pp:6594-6606
Publication Date(Web):March 18, 2013
DOI:10.1021/jp311701r
The possible formation pathways of CHx (x = 1–3) and C–C chain involved in C2 oxygenate formation from syngas on an open Cu(110) surface have been systematically investigated to identify the preference mechanism of CHx (x = 1–3) and C–C chain formation. Here, we present the main results obtained from periodic density functional calculations. Our results show that all CHx (x = 1–3) species formation starts with CHO hydrogenation; among them, CHx (x = 2, 3) are the most favored monomers, however, CH3OH is the main product from syngas on the Cu(110) surface, and the formation of CHx (x = 1–3) cannot compete with CH3OH formation. Further, on the basis of the favored monomer CHx (x = 2, 3), we probe into the C–C chain formation of C2 oxygenates by CO or CHO insertion into CHx (x = 2, 3), as well as the hydrogenation, dissociation, and coupling of CHx (x = 2, 3), suggesting that CO insertion into CH2 to form C2 oxygenates is the dominant reaction for CH2 on the Cu(110) surface with an activation barrier of 44.5 kJ·mol–1; however, for CH3, CH3 hydrogenation to CH4 is the dominant reaction on the Cu(110) surface with an activation barrier of 67.5 kJ·mol–1. As a result, to achieve high productivity and selectivity for C2 oxygenates from syngas, Cu has to get help from the promoters, which should be able to boost CH2 formation and/or suppress CH3OH and CH3 formation. The present study provides the basis to understand and develop novel Cu-based catalysts for C2 oxygenate formation from syngas.
CH4 dehydrogenation on Rh(1 1 1), Rh(1 1 0) and Rh(1 0 0) surfaces has been investigated by using density functional theory (DFT) slab calculations. On the basis of energy analysis, the preferred adsorption sites of CHx (x = 0–4) and H species on Rh(1 1 1), Rh(1 1 0) and Rh(1 0 0) surfaces are located, respectively. Then, the stable co-adsorption configurations of CHx (x = 0–3) and H are obtained. Further, the kinetic results of CH4 dehydrogenation show that on Rh(1 1 1) and Rh(1 0 0) surfaces, CH is the most abundant species for CH4 dissociation; on Rh(1 1 0) surface, CH2 is the most abundant species, our results suggest that Rh catalyst can resist the carbon deposition in the CH4 dehydrogenation. Finally, results of thermodynamic and kinetic show that CH4 dehydrogenation on Rh(1 0 0) surface is the most preferable reaction pathway in comparison with that on Rh(1 1 1) and Rh(1 1 0) surfaces.
A density-functional theory method has been conducted to investigate the dissociation of CH4 on NiCu (1 1 1) surface. Two models: uniform surface slab model (Model A) and Cu-rich surface slab model (Model B) have been constructed to represent the NiCu (1 1 1) surface, in which the ratio of Ni/Cu is unit. The obtained results on the two models have been compared with those obtained on pure Ni (1 1 1) and Cu (1 1 1). It is found that the adsorption of CHx(x = 1–3) on Model B are weaker than on Model A. The rate-determining steps of CH4 dissociation on Model A and B both are the dissociation of CH, and the corresponding activation barriers are 1.37 and 1.63 eV, respectively. Obviously, it is approximately equal on Model A to that on pure Ni (1 1 1) [H. Liu, R. Zhang, R. Yan, B. Wang, K. Xie, Applied Surface Science 257 (2011) 8955], while it is lower by 0.58 eV on Model B compared to that on pure Cu (1 1 1). Therefore, the Cu-rich surface has better carbon-resistance ability than the uniform one. Those results well explain the experimental facts that NiCu/SiO2 has excellent catalytic performance and long-term stability [H.-W. Chen, C.-Y. Wang, C.-H. Yu, L.-T. Tseng, P.-H. Liao, Catalysis Today 97 (2004) 173], however, there is serious carbon deposition on NiCu/MgO–Al2O3 in CO2 reforming of methane [J. Zhang, H. Wang, A. K. Dalai, Journal of Catalysis 249 (2007) 300].
The interaction mechanism of H2S with different Cu2O(1 1 1) surfaces, including perfect, oxygen-vacancy and sulfur-containing surfaces, have been systematically studied using periodic density functional calculations. Different kinds of possible modes of H2S, as well as the resultant SH and S species adsorbed on these surfaces are identified. Two types of pathways via molecular and dissociative adsorption processes are mapped out. Our results show that sulfur species (H2S, SH and S) interact with surface Cu centers; H2S exists in the form of molecular adsorption on perfect and sulfur-containing surfaces; the dissociative adsorption of H2S occurs predominantly on oxygen-vacancy surface, suggesting that oxygen-vacancy exhibits a strong catalytic activity toward the dissociation of H2S. On the other hand, the dissociation processes of the molecular and dissociative adsorption H2S, leading to final product S species on these Cu2O(1 1 1) surfaces, show that the overall dissociation process is exothermic. Meanwhile, with respect to molecular adsorption H2S, the activation barrier and reaction energy of the overall dissociation process on perfect and oxygen-vacancy surfaces indicate that H2S can easily dissociate into S species. Importantly, in the case of dissociative adsorption of H2S, the dissociation of H2S into S species is a spontaneous process with respect to molecular adsorption H2S. However, on sulfur-containing surface, the presence of surface S atom goes against the HS bond-breaking process both thermodynamically and kinetically. Finally, the vibrational frequencies for the adsorbed H2S, SH and S species on these surfaces have been obtained, which can be applied to guide surface vibrational spectroscopy in experiment.
A density-functional theory method has been conducted to investigate the interactions of NiM (M = Mn, Fe, Co and Cu) with MgO (1 0 0) as well as the effects of interactions on the adsorption of CO2. The binding energies of NiM on MgO and the adsorption energies of CO2 on NiM/MgO have been calculated, and the results show that the defective NiM/MgO catalysts exhibit stronger metal–support interaction (MSI) than the perfect NiM/MgO catalysts do, leading to weaker adsorption ability to CO2, except NiMn/MgO system. However, for the catalysts with the same MgO surface and different bimetals, the stronger the MSI is, the stronger adsorption ability of the substrate to CO2 is, except NiCu/MgO system.
Co-reporter:Riguang Zhang, Hongyan Liu, Baojun Wang, and Lixia Ling
The Journal of Physical Chemistry C 2012 Volume 116(Issue 42) pp:22266-22280
Publication Date(Web):October 4, 2012
DOI:10.1021/jp211900z
The mechanism of HCOOH decomposition on Pd(111) surface leading to the formation of CO2 and CO has been systematically investigated to identify the preference of CO2 or CO as the dominant product. Here, we present the main results obtained from periodic, self-consistent density functional theory calculations. Four possible pathways of HCOOH decomposition, initiated by the activation of the O–H, C–H, and C–O bonds of HCOOH, as well as the activation of simultaneous C–H and C–O bonds of HCOOH, have been proposed and discussed. Then, the effects of coadsorbed H2O and its coverage on the decomposition of HCOOH have been also considered. Our results show that CO2 is preferentially formed as the dominant product of HCOOH decomposition on Pd(111) surface via a dual-path mechanism, which involves both the carboxyl (trans-COOH) and formate (bi-HCOO) intermediates, along with alternative bond-breaking possible steps in those intermediates. The dehydrogenation of HCOOH on Pd surface is a vital process for CO2 formation. Further, the coadsorbed H2O and its coverage play an important role in the decomposition of HCOOH, and the preferred catalytic pathway of CO2 formation is qualitatively dependent on surface H2O coverage. Therefore, our results would at the microscopic level provide insights into the mechanism, energetics, and possible reactive intermediates of HCOOH decomposition regarding the preference of CO2 formation as the dominant product for the catalytic reactions involving HCOOH and for a direct HCOOH fuel cell on Pd system.
Co-reporter:Ri-Guang Zhang;Li-Xia Ling;Bao-Jun Wang
Journal of Molecular Modeling 2012 Volume 18( Issue 4) pp:1255-1262
Publication Date(Web):2012 April
DOI:10.1007/s00894-011-1155-8
The detailed mechanisms of the hydrolysis of carbonyl sulfide (OCS) by nucleophilic water and hydroxide ion in both the gas phase and bulk water solvent have been investigated using density functional theory. Various reaction channels on the potential surface have been identified. The thermodynamic results demonstrate that the hydrolysis of OCS by nucleophilic water and hydroxide ion should proceed more favorably at low temperature. The hydrolysis of OCS by the hydroxide ion is the main reaction channel from thermodynamic and kinetic perspectives, and the bulk solvent can influence the rate-determining step in this channel. However, the solvent barely modifies the activation energy of the rate-determining step. For the hydrolysis of OCS by nucleophilic water, the solvent does not modify the rate-determining step, and the corresponding activation energy of the rate-determining step barely changes. This bulk solvent effect suggests that most of the contribution of the solvent is accounted for by considering one water molecule and a hydroxide ion.
The formation mechanism of CH3O by the adsorption and decomposition of CH3OH on clean and oxygen-precovered Cu2O(1 1 1) surface has been investigated with density functional theory method together with the periodic slab models. Two possible formation pathways of CH3O by CH3OH decomposition on oxygen-precovered (Opre) Cu2O(1 1 1) surface were proposed and discussed. One is the O–H bond-cleavage of CH3OH with H migration to Opre to form CH3O; the other is the C–O bond-scission of CH3OH with CH3 migration to Opre leading to CH3Opre. The calculated results show that the O–H bond-breaking path has the lowest activation barrier 26.8 kJ mol−1, the presence of oxygen-precovered on Cu2O(1 1 1) surface exhibits a high surface reactivity toward the formation of CH3O by the O–H bond-cleavage of CH3OH, and reduce the activation barrier of O–H bond-cleavage. The C–O bond-breaking path was inhibited by dynamics, suggesting that the O atom of CH3O is not from the oxygen-precovered, but comes from the O of CH3OH. Meanwhile, the calculated results give a clear illustration about the formation mechanism of CH3O in the presence of oxygen and the role of oxygen at the microscopic level.
The adsorption and dissociation of O2 on the perfect and oxygen-deficient Cu2O(1 1 1) surface have been systematically studied using periodic density functional calculations. Different kinds of possible modes of atomic O and molecular O2 adsorbed on the Cu2O(1 1 1) surface are identified: atomic O is found to prefer threefold 3Cu site on the perfect surface and Ovacancy site on the deficient surface, respectively. CuCUS is the most advantageous site with molecularly adsorbed O2 lying flatly over singly coordinate CuCUS–CuCSA bridge on the perfect surface. O2 adsorbed dissociatively on the deficient surface, which is the main dissociation pathway of O2, and a small quantity of molecularly adsorbed O2 has been obtained. Further, possible dissociation pathways of molecularly adsorbed O2 on the Cu2O(1 1 1) surface are explored, the reaction energies and relevant barriers show that a small quantity of molecularly adsorbed O2 dissociation into two O atoms on the deficient surface is favorable both thermodynamically and kinetically in comparison with the dissociation of O2 on the perfect surface. The calculated results suggest that the presence of oxygen vacancy exhibits a strong chemical reactivity towards the dissociation of O2 and can obviously improve the catalytic activity of Cu2O, which is in agreement with the experimental observation.
A density-functional theory method has been conducted to investigate the association of C + O on (1 1 1) facets of ordered NiCo alloy and the results have been compared with those obtained on pure Ni(1 1 1) surface. In reaction of C + O, the favorable reaction path is that C adsorbed on HCP-1 site moves to the nearest Ni–Co bridge site, and associates with O migrating from FCC-1 site to result in CO adsorbed on the bridge site of Ni–Co. However, the reaction barrier is higher by 0.35 eV than that on pure Ni(1 1 1), which indicates that the incorporation of Co into the Ni crystal is not in favor of the reaction of carbon delimination.
A density-functional theory method has been conducted to investigate the adsorption of CHx (x = 0–4) as well as the dissociation of CHx (x = 1–4) on (1 1 1) facets of ordered NiCo alloy. The results have been compared with those obtained on pure Ni (1 1 1) surface. It shows that the adsorption energies of C and CH are decreased while it is increased for CH3 on NiCo (1 1 1) compared to those on pure Ni (1 1 1). Furthermore, on NiCo (1 1 1), dissociation of CHx prefers not to the top of Ni, but to the top of Co. The rate-determining step for CH4 dissociation is considered as the first step of dehydrogenation on NiCo (1 1 1), while it is the fourth step of dehydrogenation on Ni (1 1 1). Furthermore, the activation barrier in rate-determining step is slightly higher by 0.07 eV on Ni (1 1 1) than that on NiCo (1 1 1). From above results, it is important to point out that carbon is easy to form on NiCo (1 1 1) although the adsorption energy of C atom is slightly decreased compared to that on Ni (1 1 1).
Co-reporter:Riguang Zhang ; Baojun Wang ; Hongyan Liu ;Lixia Ling
The Journal of Physical Chemistry C 2011 Volume 115(Issue 40) pp:19811-19818
Publication Date(Web):September 4, 2011
DOI:10.1021/jp206065y
Catalytic hydrogenation of CO2 to methanol is a promising way to recycle and utilize CO2. In this study, the elementary steps leading to HCOO and CO formation have been explored to identify hydroxylation effect of the oxide support on the selectivity in CO2 hydrogenation on Cu/γ-Al2O3 catalyst by the density functional theory (DFT) slab calculations. Two models: Cu4 cluster supported on the dry γ-Al2O3(110) surface, D(Cu4), and on the hydroxylated γ-Al2O3(110) surface, H(Cu4), have been used to model Cu/γ-Al2O3. On D(Cu4), the formation of HCOO is preferred kinetically. On H(Cu4), HCOO formation is still kinetically favorable. These results indicate that the hydroxylation of γ-Al2O3 support cannot alter the pathway of CO2 hydrogenation forming the dominate product HCOO, and ultimately, the selectivity of CO2 hydrogenation for HCOO formation on Cu/γ-Al2O3 is higher, which supports the experimental fact that Al2O3-supported Cu catalyst is widely used to synthesize methanol by CO2 hydrogenation.
DFT calculations have been performed to investigate the effect of dielectric responses of the solvent environment on the CO adsorption over CuCl(1 1 1) surface by using COSMO (conductor-like solvent model) model in Dmol3. Different dielectric constants, including vacuum, liquid paraffin, methylene chloride, methanol and water solution, are considered. The effects of solvent model on the structural parameters, adsorption energies and vibrational frequency of CO adsorption over CuCl(1 1 1) surface have been investigated. The calculation results suggest that solvent effects can improve the stability of CO adsorption and reduce the intensity of C–O bond, which might mean that solvent is in favor of C–O bond activation and improve the reaction activity of oxidative carbonylation in a slurry reactor.
Interactions of atomic and molecular hydrogen with perfect and deficient Cu2O(1 1 1) surfaces have been investigated by density functional theory. Different kinds of possible modes of H and H2 adsorbed on the Cu2O(1 1 1) surface and possible dissociation pathways were examined. The calculated results indicate that OSUF, CuCUS and Ovacancy sites are the adsorption active centers for H adsorbed on the Cu2O(1 1 1) surface, and for H2 adsorption over perfect surface, CuCUS site is the most advantageous position with the side-on type of H2. For H2 adsorption over deficient surface, two adsorption models of H2, H2 adsorbing perpendicularly over Ovacancy site and H2 lying flatly over singly-coordinate Cu–Cu short bridge, are typical of non-energy-barrier dissociative adsorption leading to one atomic H completely inserted into the crystal lattice and the other bounded to CuCUS atom, suggesting that the dissociative adsorption of H2 is the main dissociation pathway of H2 on the Cu2O(1 1 1) surface. Our calculation result is consistent with that of the experimental observation. Therefore, Cu2O(1 1 1) surface with oxygen vacancy exhibits a strong chemical reactivity towards the dissociation of H2.
Journal of Molecular Structure: THEOCHEM 2010 Volume 952(1–3) pp:31-35
Publication Date(Web):30 July 2010
DOI:10.1016/j.theochem.2010.04.001
The migration of sulfur resulting in the formation of CS, H2S and thiophene during benzenethiol pyrolysis has been investigated using the density functional theory method with PW91 functional and DND basis set. The lowest energy path is that H of the thiol group transfers to the ipso C and S radical is eliminated by beta scission reaction, and then the S radical combines with H radical formed during coal pyrolysis and eventually results in the formation of H2S. The formation of H2S via benzenthiol pyrolysis is easier than via thiophene by comparing with the kinetic data.
Journal of Molecular Modeling 2010 Volume 16( Issue 12) pp:1911-1917
Publication Date(Web):2010 December
DOI:10.1007/s00894-010-0686-8
The reaction mechanisms of H2 with OCS have been investigated theoretically by using density function theory method. Three possible pathways leading to major products CO and H2S, as well as two possible pathways leading to by-product CH4 have been proposed and discussed. For these reaction pathways, the structure parameters, vibrational frequencies and energies for each stationary point have been calculated, and the corresponding reaction mechanism has been given by the potential energy surface, which is drawn according to the relative energies. The calculated results show that the corresponding major products CO and H2S as well as by-product CH4 are in agreement with experimental findings, which provided a new illustration and guidance for the reaction of H2 with OCS.
Journal of Molecular Structure: THEOCHEM 2009 Volume 905(1–3) pp:8-12
Publication Date(Web):15 July 2009
DOI:10.1016/j.theochem.2009.02.040
The pyrolysis mechanism of thiophene in coal has been investigated with a density functional theory method. Three intramolecular hydrogen migration reaction paths leading to the formation of H2S were designed. It can be concluded that the favorable energy path by the kinetic analysis is that the α-H migrates to S firstly; then the β-H migrates to the α-C followed by a concerted C–S bond cleavage resulting in the cyclic structure of thiophene turning into a chain structure; finally H2S and butadiyne are formed via H migrating twice. In the favorable energy path, the rate determining step is the α-H migration to S, and the activation energy is about 351.63 kJ mol−1 at 298.15 K.
Co-reporter:Lina Zhang, Lixia Ling, Senpeng Zhao, Riguang Zhang, Baojun Wang
Journal of Energy Chemistry (September 2014) Volume 23(Issue 5) pp:669-678
Publication Date(Web):1 September 2014
DOI:10.1016/S2095-4956(14)60183-2
The formation mechanism of methane (CH4) during coal evolution has been investigated by density functional theory (DFT) of quantum chemistry. Thermogenic gas, which is generated during the thermal evolution of medium rank coal, is the main source of coalbed methane (CBM). Ethylbenzene (A) and 6,7-dimethyl-5,6,7,8-tetrahydro-1-hydroxynaphthalene (B) have been used as model compounds to study the pyrolysis mechanism of highly volatile bituminous coal (R), according to the similarity of bond orders and bond lengths. All possible paths are designed for each model. It can be concluded that the activation energies for H-assisted paths are lower than others in the process of methane formation; an H radical attacking on β-C to yield CH4 is the dominant path for the formation of CH4 from highly volatile bituminous coal. In addition, the calculated results also reveal that the positions on which H radical attacks and to which intramolecular H migrates have effects on methyl cleavage.The divided models of highly volatile bituminous coal were used to investigate the formation mechanism of methane during coal evolution by density functional theory method.Download full-size image
Chinese Journal of Chemical Engineering (October 2009) Volume 17(Issue 5) pp:805-813
Publication Date(Web):1 October 2009
DOI:10.1016/S1004-9541(08)60280-3
The pyrolysis mechanisms of quinoline and isoquinoline were investigated using the density functional theory of quantum chemistry, including eight reaction paths and a common tautomeric intermediate 1-indene imine. It is concluded that the conformational tautomerism of the intermediate decides the pyrolysis products (C6H6HC≡C—C≡N, C6H5C≡N and HC≡CH) to be the same, and also decides the total disappearance rates of the reactants to be the same, for both original reactants quinoline and isoquinoline during the pyrolysis reaction. The results indicate that the intramolecular hydrogen migration is an important reaction step, which often appears in the paths of the pyrolysis mechanism. The activation energies of the rate determining steps are obtained. The calculated results are in good agreement with the experimental results.
Journal of Natural Gas Chemistry (November 2011) Volume 20(Issue 6) pp:611-617
Publication Date(Web):1 November 2011
DOI:10.1016/S1003-9953(10)60252-6
A density-functional theory (DFT) method has been conducted to systematically investigate the adsorption of CHx (x = 0∼4) as well as the dissociation of CHx (x = 1∼4) on (111) facets of gold-alloyed Ni surface. The results have been compared with those obtained on pure Ni(111) surface. It shows that the adsorption energies of CHx(x = 1∼3) are lower, and the reaction barriers of CH4 dissociation are higher in the first and the fourth steps on gold-alloyed Ni(111) compared with those on pure Ni(111). In particular, the rate-determining step for CH4 dissociation is considered as the first step of dehydrogenation on gold-alloyed Ni(111), while it is the fourth step of dehydrogenation on pure Ni(111). Furthermore, the activation barrier in rate-determining step is higher by 0.41 eV on gold-alloyed Ni(111) than that on pure Ni(111). From above results, it can be concluded that carbon is not easy to form on gold-alloyed Ni(111) compared with that on pure Ni(111).
Chinese Journal of Chemical Engineering (June 2009) Volume 17(Issue 3) pp:394-400
Publication Date(Web):1 June 2009
DOI:10.1016/S1004-9541(08)60222-0
The free-radical growth mechanisms for the formation of polycyclic arenes (PCAs) were constructed based on the block unit of benzene, and were calculated by the quantum chemistry PM3 method. Two kinds of reaction paths are proposed and discussed. The calculation results show that the formation of PCAs is only controlled by the elimination of H atom from benzene, and the corresponding activation energy is 307.60 kJ·mol−1. H2 is only the effluent gas in our proposed reaction mechanism, and the calculation results are in accord with the experimental facts.
Co-reporter:Lixia Ling, Jianbing Wu, Jiajia Song, Peide Han, Baojun Wang
Computational and Theoretical Chemistry (15 November 2012) Volume 1000() pp:
Publication Date(Web):15 November 2012
DOI:10.1016/j.comptc.2012.09.011
The adsorption and dissociation of H2S on the oxygen-deficient ZnO(101¯0) surface have been investigated by using the density functional theory (DFT) method with PW91 functional and DNP basis set. H2S is dissociatively adsorbed on the oxygen-deficient ZnO(101¯0) surface without an energy barrier, which is the first dehydrogenation step. The second dehydrogenation step takes place via H transferring from S in SH to the surface O atom forming OH group by overcoming an energy barrier of 22.69 kJ·mol−1. Our results show that the adsorption of the dissociated S atom is more stable on the oxygen-deficient surface than on the perfect surface.Graphical abstractHighlights► H2S prefers to adsorb dissociatively on the oxygen-deficient ZnO(101¯0) surface. ► H2S can easily dissociate into S species. ► The dissociated S atom fills the oxygen vacancy on the ZnO(101¯0) surface. ► The oxygen vacancy is unfavorable to the desulfurization of H2S by ZnO sorbent.
Co-reporter:Riguang Zhang, Mao Peng, Tian Duan, Baojun Wang
Applied Surface Science (15 June 2017) Volume 407() pp:
Publication Date(Web):15 June 2017
DOI:10.1016/j.apsusc.2017.02.164
•Cu13, Cu38 and Cu55 clusters are employed to model different sizes of Cu cluster.•The adsorption ability of the species decreases with the increasing of Cu cluster size.•Cu cluster size affects the abundant form of CHx(x = 1–3) species and C2 oxygenates.•The smaller Cu cluster size is, the higher the selectivity of C2 oxygenates is.•Cu13 cluster exhibits the highest selectivity toward C2 oxygenates among three Cu clusters.Size dependence of C2 oxygenate formation from syngas on Cu cluster has been investigated to qualitatively probe into the effect of Cu cluster size on the selectivity of C2 oxygenates, which includes two key steps: CHx and C2 oxygenate formations; Cu13, Cu38 and Cu55 clusters have been employed to model different sizes of Cu cluster. Here, density functional theory method has been performed. Our results show that the adsorption ability of the species involving in C2 oxygenate formation decreases with the increasing of Cu cluster size; Cu cluster size significantly affects the dominant existence forms of CHx(x = 1–3) species and C2 oxygenates. Among three Cu clusters, Cu13 cluster exhibits the highest selectivity toward C2 oxygenates compared to other two clusters, suggesting that Cu cluster size can affect the selectivity toward C2 oxygenates, moreover, the smaller Cu cluster size is, the higher the selectivity of C2 oxygenates is. That is probably related to the high concentration of low-coordinated defect sites on small size Cu cluster, which results in the higher activity and selectivity toward C2 oxygenates. The identification of higher intrinsic selectivity of C2 oxygenates, active sites, and stronger cluster size effect would be valuable for developing more efficient and stable Cu catalyst with higher selectivity toward C2 oxygenate in syngas conversion.
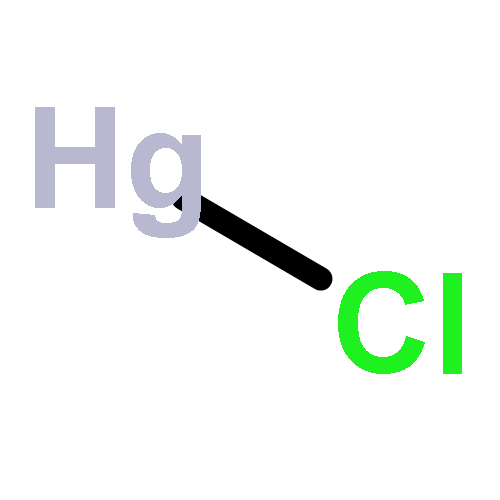
Applied Surface Science (1 May 2017) Volume 403() pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.apsusc.2017.01.126
•Mn and Fe doped CeO2(111) surfaces favor Hg capture via strong interaction.•HgS adsorbs on Mn and Fe/CeO2(111) surfaces with molecule mode.•Hg is easily oxidized by the active S on Mn and Fe/CeO2(111) surfaces.•Mn and Fe doped ceria have the potential to simultaneous remove Hg and H2S.The effects of Mn and Fe doping into the CeO2(111) surface on the simultaneous removal of Hg and H2S was investigated, a density functional theory calculation with the on-site Coulomb interaction taken into account was adopted. The adsorptions of Hg-containing species on perfect CeO2(111), Mn/CeO2(111) and Fe/CeO2(111) surfaces were studied, the results showed that Mn and Fe dopants facilitated Hg adsorption, and more charge transferred from Hg atom to the metal doped surfaces; HgS preferred to adsorb on the perfect surface with the dissociated mode, while with the molecular mode on Mn/CeO2(111) and Fe/CeO2(111) surfaces. The reaction mechanism show that the dissociated S by H2S can easily react with Hg leading to the formation of HgS on Mn/CeO2(111) and Fe/CeO2(111) surfaces, which is crucial to capture mercury.Hg and H2S can be simultaneous removed via the formation of HgS over Mn/Fe doped CeO2(111) surfaces, while Hg desorbs from the perfect surface.
Co-reporter:Xiaobin Hao, Baojun Wang, Qiang Wang, Riguang Zhang and Debao Li
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 26) pp:NaN17618-17618
Publication Date(Web):2016/05/20
DOI:10.1039/C6CP01689H
CO adsorption and activation on Ni(100), (110) and (111) surfaces have been systematically investigated to probe the effect of coverage and surface structure on CO adsorption and activation. Herein, dispersion-corrected density functional theory calculations (DFT-D) were employed, and the related thermodynamic energies at 523 K were calculated by including the zero-point energy, thermal energy and entropic corrections; the results show that the saturated coverage of CO on the Ni(111), (100) and (110) surfaces correspond to 8/9, 9/12 and 9/9 ML, respectively. As the coverage increases, the stepwise adsorption free energies decrease on the flat (111) and (100) surfaces, whereas small changes occur on the corrugated (110) surface. CO migrates from the three-fold hollow site to the top site on the (111) surface, and from the four-fold hollow to the two-fold bridge site on the (100) surface, while all the CO molecules remain at the short-bridge site on the (110) surface. As a result, the obtained intermolecular CO–CO repulsive interactions on the flat surface are stronger than the interactions on the corrugated surface. Furthermore, the computed CO vibrational frequencies at different levels of coverage over the Ni surfaces agree well with the experimental results. On the other hand, kinetic analyses were utilized to compare the stepwise CO desorption with the dissociation at different degrees of coverage on the three Ni surfaces. CO desorption is more favorable than its dissociation at all coverage levels on the most exposed Ni(111) surface. Analogously, CO desorption becomes more favorable than its dissociation on the Ni(110) surface at higher coverage, except for coverage of 1/9 ML, in which CO desorption competes with its dissociation. However, on the Ni(100) surface, CO dissociation is more favorable than its desorption at 1/12 ML; when the coverage increases from 2/12 to 3/12 ML, equilibrium states exist between dissociation and desorption over the surface; when the coverage is greater than or equal to 4/9 ML, CO desorption becomes more favorable than dissociation. By applying the atomistic thermodynamics method, the determination of stable coverage as a function of temperature and partial pressure provides useful information, not only for surface science studies under ultrahigh vacuum conditions, but also for practical applications at high temperature and pressure in exploring reactions.
Co-reporter:Lixia Ling, Zhongbei Zhao, Baojun Wang, Maohong Fan and Riguang Zhang
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 16) pp:NaN11156-11156
Publication Date(Web):2016/03/22
DOI:10.1039/C6CP01422D
The density functional theory (DFT) method has been performed to study the effects of CO and CO2 on the desulfurization of H2S over a ZnO sorbent. It shows that COS is inevitably formed on the ZnO(100) surface, which tends to be adsorbed onto the surface via a S–C bond binding with either a long or a short Zn–O bond. Potential energy profiles for the COS formation via reactions between H2S and CO, and H2S and CO2 on the ZnO(100) surface have been constructed. In the presence of CO, the dissociated active S of H2S reacting with CO leads to the formation of COS, and the activation energy of the rate-determining step is 87.7 kJ mol−1. When CO2 is present, the linear CO2 is first transferred to active CO2 in a triplet state, and then combines with active S to form COS with the highest energy barrier of 142.4 kJ mol−1. Rate constants at different temperatures show that the formation of COS via the reaction of CO and H2S is easier than that of CO2 and H2S over the ZnO surface.
The catalytic selectivity, the functions of a TiO2 support and promoter, and the mechanism of ethanol synthesis from syngas on a Rh/TiO2 model catalyst have been fully identified. Our results show that all species preferentially interact with Rh7 clusters of a Rh/TiO2 catalyst, rather than the support and cluster–support interface. CO → CHO → CH2O → CH3O is an optimal pathway. CH3 formed via the CH3O → CH3 + O route is the most favored CHx (x = 1–3) monomer, and this route is more favorable than methanol formation by CH3O hydrogenation; CO insertion into CH3 can then form CH3CO, followed by successive hydrogenation to ethanol. Methane is formed by CH3 hydrogenation. The Rh/TiO2 catalyst exhibits better catalytic activity and selectivity toward CH3 than CH3OH formation. Starting from the CH3 species, CH4 formation is more favorable than CH3CO formation; thus, ethanol productivity and selectivity on a Rh/TiO2 catalyst with a support is determined only by CH4 formation, which is similar to that on a pure Rh catalyst without a support. Introducing an Fe promoter into the Rh/TiO2 catalyst effectively suppresses methane production, and promotes CH3CO formation. Therefore, compared to a pure Rh catalyst without a support, the TiO2 support serves only to promote the activity and selectivity of CH3 formation, and provide more CH3 species for ethanol formation; methane formation is independent of the Rh catalyst support, and depends only on the promoter. In order to achieve high ethanol productivity and selectivity, an effective Rh-based catalyst must contain a suitable combination of supports and promoters, in which the promoter, M, should have characteristics that draw the d-band center of the MRh/TiO2 catalyst closer to the Fermi level compared to the Rh7/TiO2 catalyst; as a result, the MRh/TiO2 catalyst can suppress CH4 production and facilitate C2 oxygenate formation.