Co-reporter:Qiao Zhou, Can Du, Li Yang, Meiyu Zhao, Yumei Dai, and Peng Song
The Journal of Physical Chemistry A June 22, 2017 Volume 121(Issue 24) pp:4645-4645
Publication Date(Web):May 31, 2017
DOI:10.1021/acs.jpca.7b04051
The single and dual cooperated proton transfer dynamic process in the excited state of 1,5-dihydroxyanthraquinone (1,5-DHAQ) was theoretically investigated, taking solvent effects (ethanol) into account. The absorption and fluorescence spectra were simulated, and dual fluorescence exhibited, which is consistent with previous experiments. Analysis of the calculated IR and Raman vibration spectra reveals that the intramolecular hydrogen bonding interactions (O20–H21···O24 and O22–H23···O25) are strengthened following the excited proton transfer process. Finally, by constructing the potential energy surfaces of the ground state, first excited singlet state, and triplet state, the mechanism of the intramolecular proton transfer of 1,5-DHAQ can be revealed.
Co-reporter:Qiang Wei, Qiao Zhou, Meiyu Zhao, Meixia Zhang, Peng Song
Journal of Luminescence 2017 Volume 183() pp:7-12
Publication Date(Web):March 2017
DOI:10.1016/j.jlumin.2016.11.024
In the present work, using density functional theory (DFT) and time-dependent density functional theory (TDDFT) methods, we investigated the compared excited-state intramolecular proton transfer (ESIPT) mechanism of 2-acetylindan-1,3-dione (AID) in both non-polar (hexane) and polar (acetonitrile) solvents theoretically. Based on the calculation of electron density ρ(r) and Laplacian ∇2ρ(r) at the bond critical point using Atoms-In-Molecule (AIM) theory, the intramolecular hydrogen bond (O–H∙∙∙O) has been proved to be existent in the S0 state. Comparing the prime structural variations of AID involved in the intramolecular hydrogen bond, we can conclude that O–H∙∙∙O should be strengthened in the S1 state, which may facilitate the ESIPT process. Concomitantly, infrared vibrational spectra analysis further verify the stability of hydrogen bond. In good agreement with previous experimental results, AID reveals two kinds of excited-state structures (AID-enol* and AID-keto*). In addition, the role of charge transfer interaction has been addressed under the frontier molecular orbitals (MOs), which depicts the nature of electronical excited state and supports the ESIPT reaction. Our scanned potential energy curves according to variational O–H coordinate demonstrates that the proton transfer process should be more likely to occur in the S1 state due to the inappreciable potential energy barriers. In addition, due to the minute differences of potential energy barriers contrasting hexane and acetonitrile solvents in the S1 state, we believe that solvent effect could play roles in controlling excited state behaviors of AID system.The excited-state intramolecular proton transfer (ESIPT) mechanism of 2-acetylindan-1,3-dione (AID) in both non-polar (hexane) and polar (acetonitrile) solvents are theoretically investigated. Based on the calculation of electron density ρ(r) and Laplacian ∇2ρ(r) at the bond critical point using Atoms-In-Molecule (AIM) theory, the intramolecular hydrogen bond (O–H∙∙∙O) has been proved to be existent in the S0 state. Which has been proved to be strengthened in the S1 state, and facilitates the ESIPT process. Finally, solvent effect could play roles in controlling excited state behaviors of AID system, according to the scanned potential energy curves along the variational O–H coordinate.
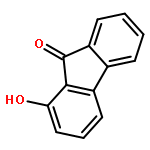
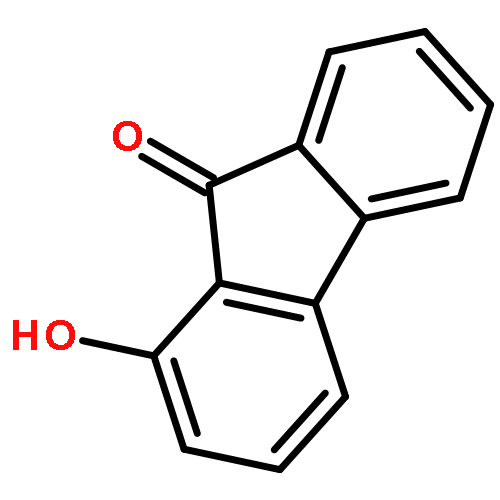
Co-reporter:Meixia Zhang;Qiao Zhou;Mengru Zhang;Yumei Dai
Journal of Cluster Science 2017 Volume 28( Issue 3) pp:1191-1200
Publication Date(Web):26 November 2016
DOI:10.1007/s10876-016-1122-8
The excited state intramolecular proton transfer (ESIPT) dynamics of the 1-hydroxy-9H-fluoren-9-one (HHF) and 1-hydroxy-11H-benzo[b]fluoren-11-one (HHBF) chromophores were investigated theoretically. The calculated bond lengths and angles, hydrogen bond energies and infrared vibrational spectra involved in the hydrogen bonding of O–H···O indicated that the intramolecular hydrogen bond was strengthened in the S1 state. Our calculated results accurately reproduced the experimental absorbance and fluorescence emission spectra, demonstrating that the adopted time-dependent density functional theory (TDDFT) method is reasonable and effective. In addition, qualitative and quantitative intramolecular charge transfer based on the frontier molecular orbitals provided the possibility of the ESIPT reaction. The potential energy curves of the ground and first excited states have been constructed to illustrate the ESIPT mechanism. Based on our calculations, we explain the equilibrium ESIPT processes observed in previous experiments.
Co-reporter:Meixia Zhang, Qiao Zhou, Can Du, Yong Ding and Peng Song
RSC Advances 2016 vol. 6(Issue 64) pp:59389-59394
Publication Date(Web):17 Jun 2016
DOI:10.1039/C6RA11140H
Based on density functional theory (DFT) and time-dependent density functional theory (TDDFT) methods, the detailed excited state intramolecular proton transfer (ESIPT) mechanism of 2,2′-dihydroxy-1,1′-naphthalazine (P.Y. 101) has been investigated theoretically. Unlike previous theoretical investigation of P.Y. 101, our calculated results not only reproduce the absorption and fluorescence spectra reported in the previous experiment, but also were completed with considering solvent effect. It further demonstrates that the TDDFT theory we adopted is very reasonable and effective. The calculations of main bond lengths and bond angles involving in the hydrogen bondings (O1–H2⋯N3 and O4–H5⋯N6) as well as the infrared vibrational spectra and as well as the calculated hydrogen bonding energies demonstrated the intramolecular hydrogen bond was strengthened in the S1 state. In addition, qualitative and quantitative intramolecular charge transfer based on the frontier molecular orbitals provided the possibility of the ESIPT reaction. The potential energy surfaces of ground state and the first excited state have been constructed to illustrate the ESIPT mechanism. Based on our calculations, the equilibrium ESIPT process exists in the S1 state. And after the radiative transition, reversed GSIPT can also occur in the S0 state.
Co-reporter:Yanling Cui, Yafei Li, Yumei Dai, Francis Verpoort, Peng Song, Lixin Xia
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2016 Volume 154() pp:130-134
Publication Date(Web):5 February 2016
DOI:10.1016/j.saa.2015.10.038
•The intramolecular hydrogen bond is strengthened in the first excited-state.•The hydrogen bond facilitates the proton transfer from hydroxyl group to the neighbor N atom.•The spontaneous excited-state intramolecular proton transfer reaction can be observed.In the present work, TDDFT has been used to investigate the excited state intramolecular proton transfer (ESIPT) mechanism of a new chromophore II [Sensors and Actuators B: Chemical. 202 (2014) 1190]. The calculated absorption and fluorescence spectra agree well with experimental results. In addition, two types of II configurations are found in the first excited state (S1), which can be ascribed to the ESIPT reaction. Based on analysis of the calculated infrared (IR) spectra of O–H stretching vibration as well as the hydrogen bonding energies, the strengthening of the hydrogen bond in the S1 state has been confirmed. The frontier molecular orbitals (MOs), Hirshfeld charge distribution and the Natural bond orbital (NBO) have also been analyzed, which displays the tendency of the ESIPT process. Finally, potential energy curves of the S0 and S1 states were constructed, demonstrating that the ESIPT reaction can be facilitated based on the photo-excitation.
Co-reporter:Jinfeng Zhao, Junsheng Chen, Yanling Cui, Jing Wang, Lixin Xia, Yumei Dai, Peng Song and Fengcai Ma
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 2) pp:1142-1150
Publication Date(Web):17 Nov 2014
DOI:10.1039/C4CP04135F
Two excited state proton transfer mechanisms of 3-hydroxyisoquinoline (3HIQ) in cyclohexane and acetic acid (ACID) were investigated based on the time-dependent density functional theory (TDDFT), suggesting a different double-proton transfer mechanism from the one proposed previously (J. Phys. Chem. B, 1998, 102, 1053). Instead of the formation of keto–enol complexes for 3HIQ self-association in cyclohexane, our theoretical results predicted that 3HIQ self-association exists in two forms: the normal form (enol/enol) and the tautomer form (keto/keto) in cyclohexane. A high barrier (37.023 kcal mol−1) between the 3HIQ enol monomer and 3HIQ keto monomer form indicated that the 3HIQ keto monomer in the ground state should not exist. In addition, the constructed potential energy surfaces of the ground state and excited state have been used to explain the proton transfer process. Upon optical excitation, the enol/enol form is excited to the first excited state, then transfers one proton, in turn, transition to the ground state to transfer another proton. A relatively low barrier (8.98 kcal mol−1) demonstrates two stable structures in the ground state. In view of the acetic acid solvent effect, two protons of 3HIQ/ACID transfer along the dihydrogen bonds in the first excited state, which is a different transfer mechanism to 3HIQ self-association. In addition, the proton transfer process provides a possible explanation for the fluorescence quenching observed.
Co-reporter:Meng Zhou, Jinfeng Zhao, Yanling Cui, Qianyu Wang, Yumei Dai, Peng Song, Lixin Xia
Journal of Luminescence 2015 Volume 161() pp:1-6
Publication Date(Web):May 2015
DOI:10.1016/j.jlumin.2014.12.049
•The hydrogen bond between the hydroxyl and phenanthrene is strengthened.•The hydrogen bond facilitates the proton transfer from the hydroxyl group to the N atom.•The spontaneous excited-state intramolecular proton transfer reaction can be observed.The dynamics of the excited-state intramolecular proton-transfer (ESIPT) reaction of 10-hydroxybenzoquinoline (HBQ) in different solvents, have been investigated based on the time-dependent density functional theory (TD-DFT) in detail. Upon excitation, the intramolecular hydrogen bond between the hydroxyl and phenanthrene functionality is significantly strengthened in the S1 state, which can be used as a reasonable tendency for facilitating the ESIPT process. In addition, the calculated vertical excitation energies in the S0 state and S1 state reproduce the experimental UV–vis absorbance and fluorescence emission spectra well. Through calculating the fluorescence spectra of the HBQ chromophore, two outcomes for this chromophore were found in the S1 state, which demonstrates that the ESIPT process occurs. The potential energy curves have been calculated to account for the mechanism of the proton-transfer process in the excited-state. As a result, the barrierless ESIPT process can occur in the S1 state with proton transfer from the O atom to the N atom. And maybe the ESIPT process is easier in methanol solvent due to the higher potential energy difference.
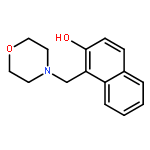
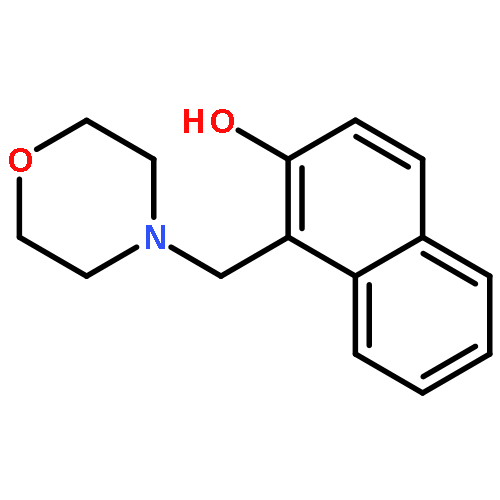
Co-reporter:Yanling Cui, Hong Zhao, Lai Jiang, Pengyu Li, Yong Ding, Peng Song, Lixin Xia
Computational and Theoretical Chemistry 2015 Volume 1074() pp:125-130
Publication Date(Web):15 December 2015
DOI:10.1016/j.comptc.2015.10.020
•The intramolecular hydrogen bond is strengthened in the first excited-state.•The hydrogen bond facilitates the proton transfer from hydroxyl group to the neighbor N atom.•The spontaneous excited-state intramolecular proton transfer reaction can be observed.In the present work, the solvent dependent excited-state intramolecular proton transfer (ESIPT) dynamics of 1-morpholinylmethyl-2-naphthol (MN) in n-hexane and acetonitrile have been investigated, based on the time-dependent density functional theory, as well as IEFPCM model. The theoretical results obtained, including the primary bond lengths, bond angles, and IR vibrational spectra, predict the possibility of ESIPT in the S1 state for MN chromophore in n-hexane and acetonitrile. In the polar solvent, all the calculated results are consistent with the experimentally observed phenomena and the dual fluorescence emission mechanism is well explained. Furthermore, our theoretical study provides the understanding for the undetected fluorescence peak at about 389 nm of the MN chromophore in n-hexane, as the fluorescence emission from keto-MN product of post-ESIPT process. Upon optical excitation, the normal enol form is excited to the first excited state, following which the proton may be transferred along the route (O17–H⋯N) with a relative large energy barrier (1.93 kcal/mol) and generate the keto tautomer. However, the low energy barrier (0.26 kcal/mol) for proton back-transfer facilitates the conformation transition from the keto to enol form.
Co-reporter:Yumei Dai, Jinfeng Zhao, Yanling Cui, Qianyu Wang, Peng Song, Fengcai Ma, Yangyang Zhao
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2015 144() pp: 76-80
Publication Date(Web):
DOI:10.1016/j.saa.2015.02.098
Co-reporter:Jiarui Xia, Meng Zhou, Shaowu Sun, Guan Wang, Peng Song, Meihua Ge
Dyes and Pigments 2014 Volume 103() pp:71-75
Publication Date(Web):April 2014
DOI:10.1016/j.dyepig.2013.11.025
•The photophysical properties of DMANF have been theoretically investigated.•The calculated absorption and fluorescence spectrum agree well with the experiment.•TICT process is responsible for the excited state fluorescence in DMANF molecule.•The nitrogen lone pair electrons play an important role in the TICT process.The ground-state and excited-state electronic structures, relaxation dynamics as well as photophysical properties of the donor–π–acceptor compound 2-dimethylamino-7-nitrofluorene (DMANF) have been theoretically investigated using density functional theory (DFT) and time-dependent density functional theory (TDDFT) methods. The calculated absorption and fluorescence spectrum agree well with the experimental results. The qualitative potential energy curves of twisting either donor or acceptor group favor the excited state intramolecular charge-transfer (ICT) relaxation process. However, the frontier molecular orbital picture supports that the twisting along the donor part is responsible for the fluorescence of the excited state. Further investigation of the strong dipolar stabilization with different structures indicates that two processes are involved in the whole twist and charge transfer dynamics: first, charge transfer is rapidly caused by the excitation to the first singlet state; second, charge transfer goes on slowly together with the twist motion. For strong dipolar stabilization, induced by twisting of the dimethylamino group, the charge transfer excited state corresponds to a more stable state with the twisted structure. Which results in these kinds of D–π–A compounds do not twist in nonpolar solvent. In the twisting charge transfer process, the nitrogen lone pair electrons play an important role.
Co-reporter:Jinfeng Zhao, Peng Song, Yanling Cui, Xuemei Liu, Shaowu Sun, Siyao Hou, Fengcai Ma
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2014 Volume 131() pp:282-287
Publication Date(Web):15 October 2014
DOI:10.1016/j.saa.2014.04.116
•Intermolecular hydrogen bond is strengthened in excited state.•Multiple hydrogen bond enhances the hydrogen bond binding energy.•Three kinds of binding sites and seven kinds of binding modes are reported.In the present work, the time-dependent density functional theory (TD-DFT) method was adopted to investigate the excited state hydrogen-bond dynamics of 2-aminopyridine monomer (2AP) and its derivatives in hydrogen donating methanol solvent. The calculated steady-state absorption and fluorescence spectra agree well with the experimental results. Theoretical results state that the bond lengths of both O–H and N–H bands are lengthened, while the intermolecular hydrogen bond lengths are shortened in the excited state. Further, the intermolecular hydrogen bonds are proved to be strengthened according to the calculated binding energy. As a reasonable explanation, the hydrogen bonds binding energy increases with multiple hydrogen-bonding interactions in the electronically excited state. In addition, the hydrogen bonding dynamics in the excited state were visualized by the spectral shifts of vibrational modes. The calculated infrared spectra of both O–H and N–H stretching vibrational regions revealed that the O–H and N–H stretching bands red-shift.Graphical abstract
Co-reporter:Jing Liu;Jiarui Xia; Peng Song; Yong Ding;Yanling Cui;Xuemei Liu; Yumei Dai; Fengcai Ma
ChemPhysChem 2014 Volume 15( Issue 12) pp:2626-2633
Publication Date(Web):
DOI:10.1002/cphc.201402026
Abstract
The ground- and excited-state properties of benzene-linked bisphenalenyl (B-LBP), naphthaline-linked bisphenalenyl (N-LBP), and anthracene-linked bisphenalenyl (A-LBP) Kekulé molecules and their respective one-dimensional (1D) stacks are investigated using time-dependent density functional theory (TD-DFT) and a range of extensive multidimensional visualization techniques. The results reveal a covalent π–π bonding interaction between overlapping phenalenyl radicals whose bond length is shorter than the van der Waals distance between carbon atoms. Increasing the linker length and/or number of molecules involved in the 1D stack decreases the HOMO–LUMO energy gap and increases the wavelength of the systems. The charge-transfer mechanism and electron coherence both differ with changes in the linker length and/or number of molecules involved in the 1D stack.
Co-reporter:Lixin Xia, Yafei Li, Jing Wang, Caiqing Ma, Peng Song
Sensors and Actuators B: Chemical (June 2017) Volume 244() pp:767-770
Publication Date(Web):June 2017
DOI:10.1016/j.snb.2017.01.090
Co-reporter:Jinfeng Zhao, Junsheng Chen, Yanling Cui, Jing Wang, Lixin Xia, Yumei Dai, Peng Song and Fengcai Ma
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 2) pp:NaN1150-1150
Publication Date(Web):2014/11/17
DOI:10.1039/C4CP04135F
Two excited state proton transfer mechanisms of 3-hydroxyisoquinoline (3HIQ) in cyclohexane and acetic acid (ACID) were investigated based on the time-dependent density functional theory (TDDFT), suggesting a different double-proton transfer mechanism from the one proposed previously (J. Phys. Chem. B, 1998, 102, 1053). Instead of the formation of keto–enol complexes for 3HIQ self-association in cyclohexane, our theoretical results predicted that 3HIQ self-association exists in two forms: the normal form (enol/enol) and the tautomer form (keto/keto) in cyclohexane. A high barrier (37.023 kcal mol−1) between the 3HIQ enol monomer and 3HIQ keto monomer form indicated that the 3HIQ keto monomer in the ground state should not exist. In addition, the constructed potential energy surfaces of the ground state and excited state have been used to explain the proton transfer process. Upon optical excitation, the enol/enol form is excited to the first excited state, then transfers one proton, in turn, transition to the ground state to transfer another proton. A relatively low barrier (8.98 kcal mol−1) demonstrates two stable structures in the ground state. In view of the acetic acid solvent effect, two protons of 3HIQ/ACID transfer along the dihydrogen bonds in the first excited state, which is a different transfer mechanism to 3HIQ self-association. In addition, the proton transfer process provides a possible explanation for the fluorescence quenching observed.

![2,9-Dibutylisoquinolino[4',5',6':6,5,10]anthra[2,1,9-def]isoquino line-1,3,8,10(2H,9H)-tetrone](http://img.cochemist.com/ccimg/52100/52000-75-6.png)
![2,9-Dibutylisoquinolino[4',5',6':6,5,10]anthra[2,1,9-def]isoquino line-1,3,8,10(2H,9H)-tetrone](http://img.cochemist.com/ccimg/52100/52000-75-6_b.png)
![BENZ[5,6]ACENAPHTHO[1,2-K]BENZO[CD]FLUORANTHENE, 6,14-DIPHENYL-](http://img.cochemist.com/ccimg/872100/872056-55-8.png)
![BENZ[5,6]ACENAPHTHO[1,2-K]BENZO[CD]FLUORANTHENE, 6,14-DIPHENYL-](http://img.cochemist.com/ccimg/872100/872056-55-8_b.png)
![2-hydroxynaphthalene-1-carbaldehyde [(2-hydroxy-1-naphthyl)methylene]hydrazone](http://img.cochemist.com/ccimg/2400/2387-03-3.png)
![2-hydroxynaphthalene-1-carbaldehyde [(2-hydroxy-1-naphthyl)methylene]hydrazone](http://img.cochemist.com/ccimg/2400/2387-03-3_b.png)