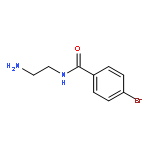
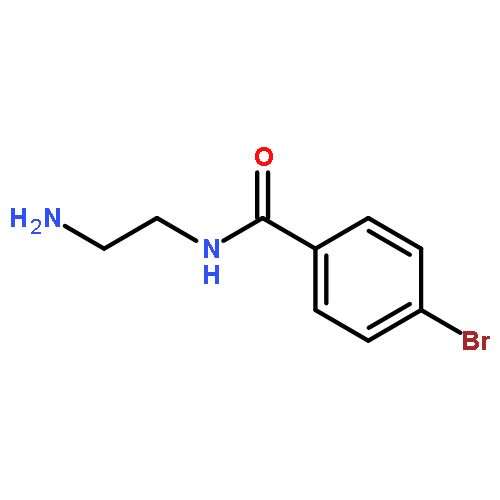
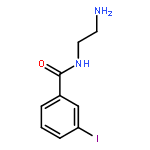
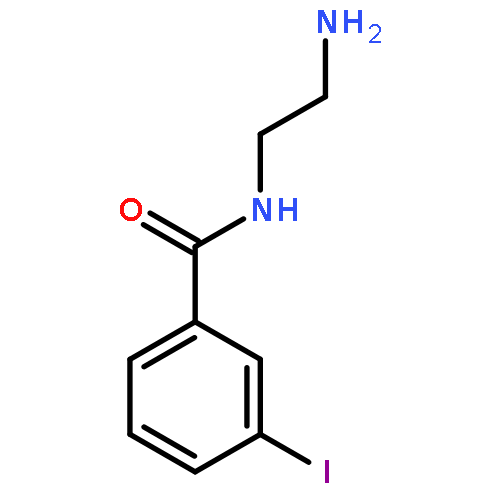
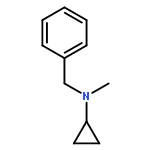
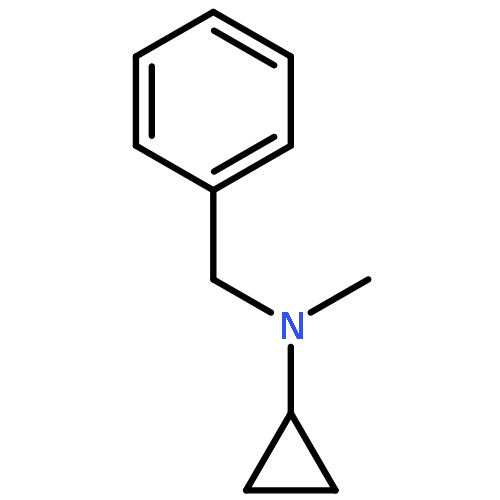
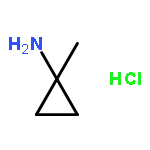
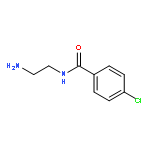
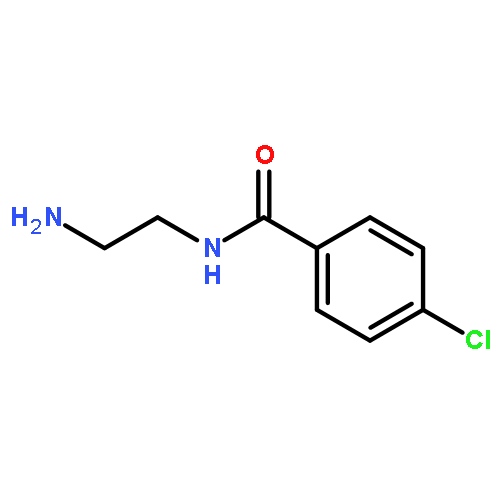
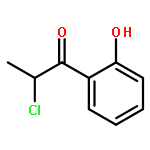
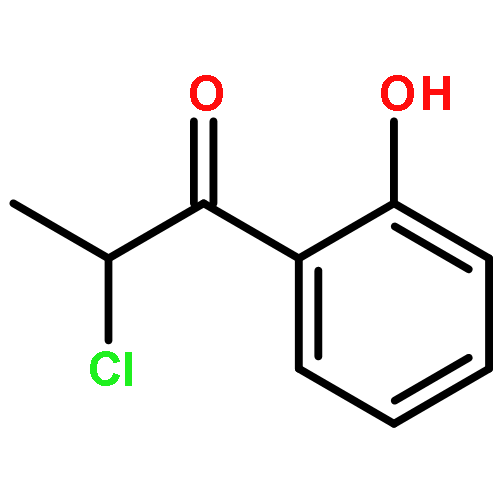
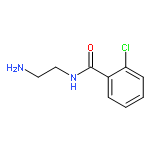
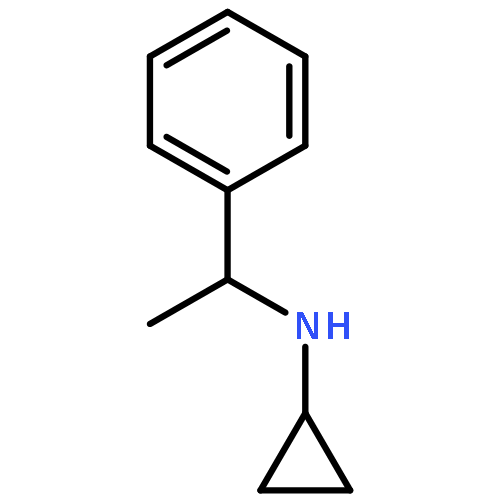
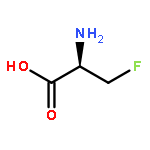
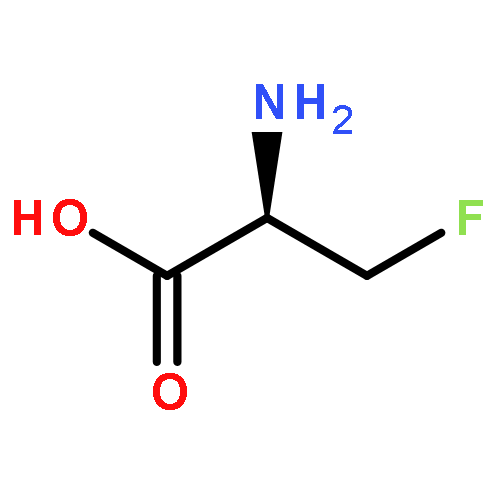
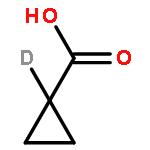
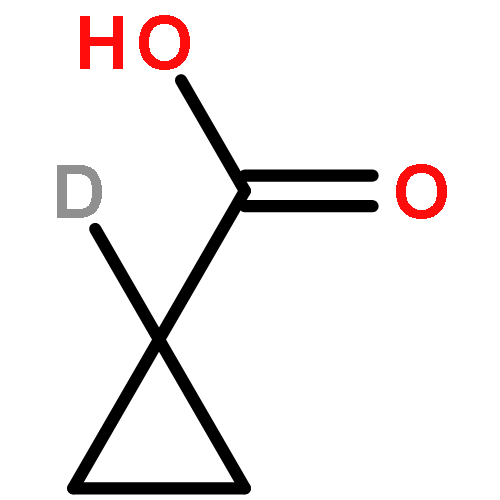
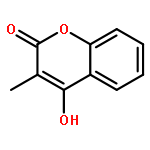
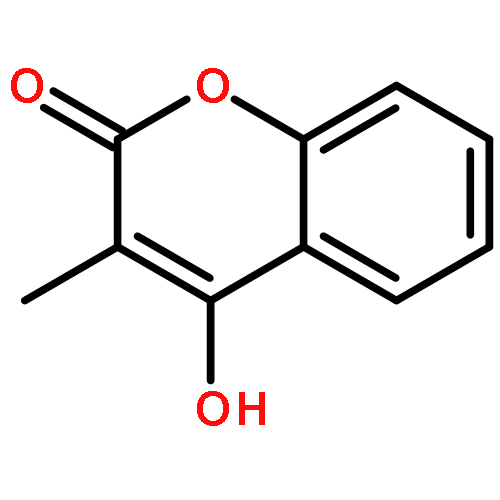
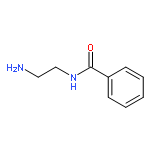
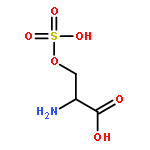
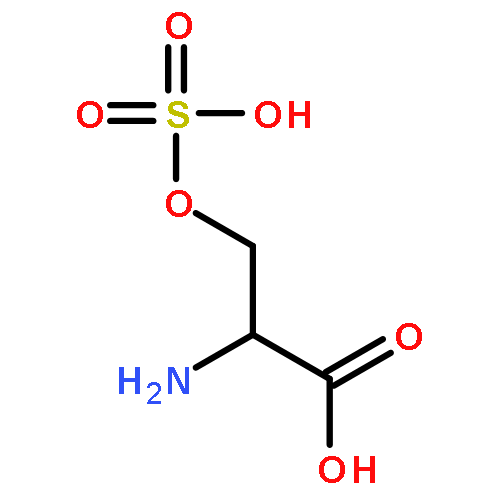
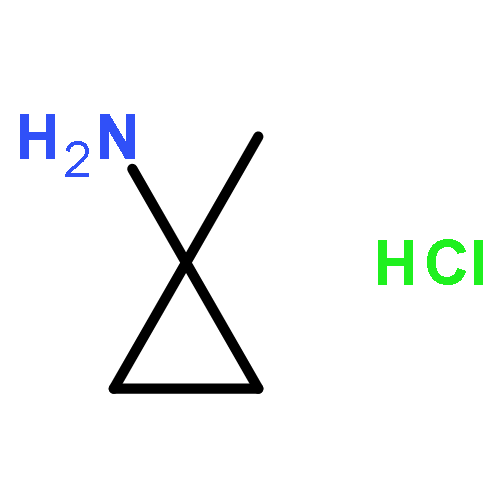Co-reporter:Romila Mascarenhas, Hoang V. Le, Kenneth D. Clevenger, Helaina J. Lehrer, Dagmar Ringe, Neil L. Kelleher, Richard B. Silverman, and Dali Liu
Biochemistry September 19, 2017 Volume 56(Issue 37) pp:4951-4951
Publication Date(Web):August 17, 2017
DOI:10.1021/acs.biochem.7b00499
Potent mechanism-based inactivators can be rationally designed against pyridoxal 5′-phosphate (PLP)-dependent drug targets, such as ornithine aminotransferase (OAT) or γ-aminobutyric acid aminotransferase (GABA-AT). An important challenge, however, is the lack of selectivity toward other PLP-dependent, off-target enzymes, because of similarities in mechanisms of all PLP-dependent aminotransferase reactions. On the basis of complex crystal structures, we investigate the inactivation mechanism of OAT, a hepatocellular carcinoma target, by (1R,3S,4S)-3-amino-4-fluorocyclopentane-1-carboxylic acid (FCP), a known inactivator of GABA-AT. A crystal structure of OAT and FCP showed the formation of a ternary adduct. This adduct can be rationalized as occurring via an enamine mechanism of inactivation, similar to that reported for GABA-AT. However, the crystal structure of an off-target, PLP-dependent enzyme, aspartate aminotransferase (Asp-AT), in complex with FCP, along with the results of attempted inhibition assays, suggests that FCP is not an inactivator of Asp-AT, but rather an alternate substrate. Turnover of FCP by Asp-AT is also supported by high-resolution mass spectrometry. Amid existing difficulties in achieving selectivity of inactivation among a large number of PLP-dependent enzymes, the obtained results provide evidence that a desirable selectivity could be achieved, taking advantage of subtle structural and mechanistic differences between a drug-target enzyme and an off-target enzyme, despite their largely similar substrate binding sites and catalytic mechanisms.
Co-reporter:Huiying Li, Heng-Yen Wang, Soosung Kang, Richard B. Silverman, and Thomas L. Poulos
Biochemistry 2016 Volume 55(Issue 26) pp:3702-3707
Publication Date(Web):June 2, 2016
DOI:10.1021/acs.biochem.6b00261
Development of potent and isoform selective nitric oxide synthase (NOS) inhibitors is challenging because of the structural similarity in the heme active sites. One amino acid difference between NOS isoforms, Asp597 in rat neuronal NOS (nNOS) versus Asn368 in bovine endothelial NOS (eNOS), has been identified as the structural basis for why some dipeptide amide inhibitors bind more tightly to nNOS than to eNOS. We now have found that the same amino acid variation is responsible for substantially different binding modes and affinity for a new class of aminopyridine-based inhibitors.
Co-reporter:Wei Tang, Huiying Li, Thomas L. Poulos, and Richard B. Silverman
Biochemistry 2015 Volume 54(Issue 15) pp:2530-2538
Publication Date(Web):March 26, 2015
DOI:10.1021/acs.biochem.5b00135
Nitric oxide synthase (NOS) catalyzes the conversion of l-arginine to l-citrulline and nitric oxide. N5-(1-Iminoethyl)-l-ornithine (l-NIO), an amidine-containing molecule, is a natural product known to be an inactivator of inducible NOS (iNOS). Because of the presence of the amidine methyl group in place of the guanidine amino group of substrate l-arginine, the active site heme peroxy intermediate sometimes cannot be protonated, thereby preventing its conversion to the heme oxo intermediate; instead, a heme oxygenase-type mechanism occurs, leading to conversion of the heme to biliverdin. This might be a new and general inactivation mechanism for heme-containing enzymes. In the studies described here, we attempted to provide support for amidines as substrates and inactivators of iNOS by the design and synthesis of amidine analogues of l-NIO having groups other than the amidine methyl group. No nitric oxide- or enzyme-catalyzed products could be detected by incubation of these amidines with iNOS. Although none of the l-NIO analogues acted as substrates, they all inhibited iNOS; increased inhibitory potency correlated with decreased substituent size. Computer modeling and molecular dynamics simulations were run on 10 and 11 to rationalize why these compounds do not act as substrates. Unlike the methyl amidine (l-NIO), the other alkyl groups block binding of O2 at the heme iron. Compounds 8, 9, and 11 were inactivators; however, no heme was lost, and no biliverdin was formed. No kinetic isotope effect on inactivation was observed with perdeuterated ethyl 8. A small amount of dimer disruption occurred with these inactivators, although the amount would not account for complete enzyme inactivation. The l-NIO analogues inactivate iNOS by a yet unknown mechanism; however, it is different from that of l-NIO, and the inactivation mechanism previously reported for l-NIO appears to be unique to methyl amidines.
Co-reporter:P. Hande Özdinler and Richard B. Silverman
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 11) pp:1179
Publication Date(Web):October 8, 2014
DOI:10.1021/ml500404b
Amyotrophic lateral sclerosis (ALS) is one of the most complex neurodegenerative diseases, involving both cortical and spinal components of motor neuron circuitry and non-neuronal cells that support the motor neurons. There is no effective therapeutic for ALS, and compounds that have extended the lifespan of ALS mouse models have failed in clinical trials. This viewpoint discusses current information regarding the changing views about ALS and what the failures in clinical trials can teach us in the search for an effective treatment. Previous challenges and roadblocks in drug discovery for ALS are noted, and solutions to current limitations are discussed. Learning from the past and moving forward with a new mindset can translate into successful and effective treatment strategies in ALS and other related diseases.
Co-reporter:Soosung Kang, Garry Cooper, Sara Fernandez Dunne, Chi-Hao Luan, D. James Surmeier, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 14) pp:4365-4373
Publication Date(Web):15 July 2013
DOI:10.1016/j.bmc.2013.04.054
The L-type calcium channel (LTCC) CaV1.3 is regarded as a new potential therapeutic target for Parkinson’s disease. Calcium influx through CaV1.3 LTCC during autonomous pacemaking in adult dopaminergic neurons of the substantia nigra pars compacta is related to the generation of mitochondrial oxidative stress in animal models. Development of a CaV1.3 antagonist selective over CaV1.2 is essential because CaV1.2 pore-forming subunits are the predominant form of LTCCs and are abundant in the central nervous and cardiovascular systems. We have explored 1,4-dihydropyrimidines and 4H-pyrans to identify potent and selective antagonists of CaV1.3 relative to CaV1.2 LTCCs. A library of 36 dihydropyridine (DHP)-mimic 1,4-dihydropyrimidines and 4H-pyrans was synthesized, and promising chiral compounds were resolved. The antagonism studies of CaV1.3 and CaV1.2 LTCCs using DHP mimic compounds showed that dihydropyrimidines and 4H-pyrans are effective antagonists of DHPs for CaV1.3 LTCCs. Some 1,4-dihydropyrimidines are more selective than isradipine for CaV1.3 over CaV1.2, shown here by both calcium flux and patch-clamp electrophysiology experiments, where the ratio of antagonism is around 2–3. These results support the hypothesis that the modified hydrogen bonding donor/acceptors in DHP-mimic dihydropyrimidines and 4H-pyrans can interact differently with DHP binding sites, but, in addition, the data suggest that the binding sites of DHP in CaV1.3 and CaV1.2 LTCCs are very similar.
Co-reporter:Jose I. Juncosa Jr., Andrew P. Groves, Guoyao Xia, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 4) pp:903-911
Publication Date(Web):15 February 2013
DOI:10.1016/j.bmc.2012.12.009
We have synthesized three analogues of 4-amino-5-fluorohexanoic acids as potential inactivators of γ-aminobutyric acid aminotransferase (GABA-AT), which were designed to combine the potency of their shorter chain analogue, 4-amino-5-fluoropentanoic acid (AFPA), with the greater enzyme selectivity of the antiepileptic vigabatrin (Sabril®). Unexpectedly, these compounds failed to inactivate or inhibit the enzyme, even at high concentrations. On the basis of molecular modeling studies, we propose that the GABA-AT active site has an accessory binding pocket that accommodates the vinyl group of vigabatrin and the fluoromethyl group of AFPA, but is too narrow to support the extra width of the distal methyl group in the synthesized analogues.
Co-reporter:Soo Hyuk Choi, Luisa Quinti, Aleksey G. Kazantsev, Richard B. Silverman
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 8) pp:2789-2793
Publication Date(Web):15 April 2012
DOI:10.1016/j.bmcl.2012.02.089
Inhibition of sirtuin 2 (SIRT2) is known to be protective against the toxicity of disease proteins in Parkinson’s and Huntington’s models of neurodegeneration. Previously, we developed SIRT2 inhibitors based on the 3-(N-arylsulfamoyl)benzamide scaffold, including3-(N-(4-bromophenyl)sulfamoyl)-N-(4-bromophenyl)benzamide(C2–8, 1a), which demonstrated neuroprotective effects in a Huntington’s mouse model, but had low potency of SIRT2 inhibition. Here we report that N-methylation of 1a greatly increases its potency and results in excellent selectivity for SIRT2 over SIRT1 and SIRT3 isoforms. Structure–activity relationships observed for 1a analogs and docking simulation data suggest that the para-substituted amido moiety of these compounds could occupy two potential hydrophobic binding pockets in SIRT2. These results provide a direction for the design of potent drug-like SIRT2 inhibitors.
Co-reporter:Fengtian Xue, Jinwen Huang, Haitao Ji, Jianguo Fang, Huiying Li, Pavel Martásek, Linda J. Roman, Thomas L. Poulos, Richard B. Silverman
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 17) pp:6526-6537
Publication Date(Web):1 September 2010
DOI:10.1016/j.bmc.2010.06.074
Selective inhibitors of neuronal nitric oxide synthase (nNOS) have the potential to develop into new neurodegenerative therapeutics. Recently, we described the discovery of novel nNOS inhibitors (1a and 1b) based on a cis-pyrrolidine pharmacophore. These compounds and related ones were found to have poor blood–brain barrier permeability, presumably because of the basic nitrogens in the molecule. Here, a series of monocationic compounds was designed on the basis of docking experiments using the crystal structures of 1a,b bound to nNOS. These compounds were synthesized and evaluated for their ability to inhibit neuronal nitric oxide synthase. Despite the excellent overlap of these compounds with 1a,b bound to nNOS, they exhibited low potency. This is because they bound in the nNOS active site in the normal orientation rather than the expected flipped orientation used in the computer modeling. The biphenyl or phenoxyphenyl tail is disordered and does not form good protein–ligand interactions. These studies demonstrate the importance of the size and rigidity of the side chain tail and the second basic amino group for nNOS binding efficiency and the importance of the hydrophobic tail for conformational orientation in the active site of nNOS.
Co-reporter:Fengtian Xue, Jianguo Fang, William W. Lewis, Pavel Martásek, Linda J. Roman, Richard B. Silverman
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 2) pp:554-557
Publication Date(Web):15 January 2010
DOI:10.1016/j.bmcl.2009.11.086
Recently, a series of potent and selective neuronal nitric oxide synthase inhibitors containing two basic nitrogen atoms was reported (Ji, H.; Stanton, B. Z.; Igarashi, J.; Li, H.; Martásek, P.; Roman, L. J.; Poulos, T. L.; Silverman, R. B. J. Am. Chem. Soc. 2008, 130, 3900–3914). In an effort to improve their bioavailability, three compounds (2a–c) were designed with electron-withdrawing groups near one of the basic nitrogen atoms to lower its pKa. Inhibition studies with these compounds showed that two of them not only retained most of the potency and selectivity of the best analogue of the earlier series, but also showed improved membrane permeability based on data from a cell-based assay.
.jpg)