Co-reporter:Dr. Eiichi Kayahara;Rui Qu; Dr. Shigeru Yamago
Angewandte Chemie 2017 Volume 129(Issue 35) pp:10564-10568
Publication Date(Web):2017/08/21
DOI:10.1002/ange.201704982
AbstractBromination of [n]cycloparaphenylenes (CPPs) is herein reported. Small [n]CPPs (n<8) underwent a bis-bromine addition reaction with high site selectively to produce tetrabromo adducts in moderate to excellent yields. Theoretical calculations revealed that thermodynamic stability dictates both the reactivity and site selectivity of the reaction. The addition product was further converted into the octabromo product by a FeBr3-catalyzed site-selective bromination reaction. The tetra- and octabromine adducts were then transformed into mono- to tetrabromo CPPs, which were further converted into several CPP derivatives. Therefore, bromination and subsequent transformations provide a path for late-stage functionalization of CPPs.
Co-reporter:Dr. Eiichi Kayahara;Rui Qu; Dr. Shigeru Yamago
Angewandte Chemie International Edition 2017 Volume 56(Issue 35) pp:10428-10432
Publication Date(Web):2017/08/21
DOI:10.1002/anie.201704982
AbstractBromination of [n]cycloparaphenylenes (CPPs) is herein reported. Small [n]CPPs (n<8) underwent a bis-bromine addition reaction with high site selectively to produce tetrabromo adducts in moderate to excellent yields. Theoretical calculations revealed that thermodynamic stability dictates both the reactivity and site selectivity of the reaction. The addition product was further converted into the octabromo product by a FeBr3-catalyzed site-selective bromination reaction. The tetra- and octabromine adducts were then transformed into mono- to tetrabromo CPPs, which were further converted into several CPP derivatives. Therefore, bromination and subsequent transformations provide a path for late-stage functionalization of CPPs.
Co-reporter:Yasuyuki Nakamura, Tasuku Ogihara, and Shigeru Yamago
ACS Macro Letters 2016 Volume 5(Issue 2) pp:248
Publication Date(Web):January 28, 2016
DOI:10.1021/acsmacrolett.5b00947
The mechanism of the Cu(I)/Cu(0)-mediated reductive coupling reactions of bromine-terminated polymer chain-end radicals, so-called atom-transfer radical coupling (ATRC), is studied. Poly(methyl acrylate) (PMA), poly(methyl methacrylate) (PMMA), and polystyrene (PSt), prepared by atom-transfer radical polymerization (ATRP), were activated by an excess amount of Cu(I)Br and Cu(0) in the presence of tris[2-(dimethylamino)ethyl]amine (Me6TREN), and the structural analyses of the resulting polymer products and deuterium-labeling experiments unambiguously determined the mechanism. While PMMA and PSt reacted by a radical mechanism as expected, PMA-bromide was reduced to an anionic species, which was most likely an organocopper species. Trapping experiments with TEMPO suggested that the polymer chain-end radicals were generated in all cases by the reduction of the bromine-terminated polymers by low-valent Cu species. However, the PMA chain-end radical was further reduced to the anionic species from which coupling products formed in low yield.
Co-reporter:Yasuyuki Nakamura;Richmond Lee;Michelle L. Coote
Macromolecular Rapid Communications 2016 Volume 37( Issue 6) pp:506-513
Publication Date(Web):
DOI:10.1002/marc.201500677
Co-reporter:Dr. Eiichi Kayahara;Kei Fukayama; Tohru Nishinaga;Dr. Shigeru Yamago
Chemistry – An Asian Journal 2016 Volume 11( Issue 12) pp:1793-1797
Publication Date(Web):
DOI:10.1002/asia.201600582
Abstract
The oxidation processes of [n]cycloparaphenylenes ([n]CPPs) (n=5–12) were systematically investigated by cyclic and rotating disk electrode voltammetry. All CPPs underwent pseudo-reversible two-electron oxidation irrespective of ring size, forming the corresponding radical cations and then dications. The results were in sharp contrast to those observed for linear oligoparaphenylenes, which only undergo one-electron oxidation. The difference in the first and second oxidation potentials in the CPP oxidation was affected by the ring size and became more significant as the decrease of CPP size. In other words, while the first oxidation from neutral CPP to the radical cation occurred faster as the size of CPP becomes smaller, the second oxidation from the radical cation to dication exhibited opposite size dependence.
Co-reporter:Dr. Eiichi Kayahara;Dr. Vijay Kumar Patel;Audrey Mercier;Dr. E. Peter Kündig;Dr. Shigeru Yamago
Angewandte Chemie 2016 Volume 128( Issue 1) pp:310-314
Publication Date(Web):
DOI:10.1002/ange.201508003
Abstract
Mono- and multinuclear complexes of ruthenium and [n]cycloparaphenylene (CPP, n=5 and 6) were synthesized in excellent yields through ligand exchange of the cationic complex [(Cp)Ru(CH3CN)3](PF6) with CPP. In the multinuclear complexes, ruthenium selectively coordinated to alternate paraphenylene units to give bis- and tris-coordinated Ru complexes for [5] and [6]CPPs, respectively. Single-crystal X-ray analysis revealed the Ru was coordinated with η6-hapticity on the convex surface of CPP.
Co-reporter:Dr. Eiichi Kayahara;Dr. Vijay Kumar Patel;Audrey Mercier;Dr. E. Peter Kündig;Dr. Shigeru Yamago
Angewandte Chemie International Edition 2016 Volume 55( Issue 1) pp:302-306
Publication Date(Web):
DOI:10.1002/anie.201508003
Abstract
Mono- and multinuclear complexes of ruthenium and [n]cycloparaphenylene (CPP, n=5 and 6) were synthesized in excellent yields through ligand exchange of the cationic complex [(Cp)Ru(CH3CN)3](PF6) with CPP. In the multinuclear complexes, ruthenium selectively coordinated to alternate paraphenylene units to give bis- and tris-coordinated Ru complexes for [5] and [6]CPPs, respectively. Single-crystal X-ray analysis revealed the Ru was coordinated with η6-hapticity on the convex surface of CPP.
Co-reporter:Eiichi Kayahara; Takahiko Kouyama; Tatsuhisa Kato
Journal of the American Chemical Society 2015 Volume 138(Issue 1) pp:338-344
Publication Date(Web):December 17, 2015
DOI:10.1021/jacs.5b10855
Radical cations and dications of [n]cyclo-p-phenylenes ([n]CPPs, n = 5, 6, 10, and 12), which are the models of those of linear oligo-p-phenylenes without a terminus, were synthesized as hexafluoroantimonate salts by the one- and two-electron chemical oxidation of CPP by NOSbF6 or SbF5. The radical cations, [n]CPP•+, and dications, [n]CPP2+, exhibited remarkable bathochromic shifts in their UV–vis–NIR absorption bands, suggesting that [n]CPP•+ and larger [n]CPP2+ exhibit longer polyene character than the shorter analogues. The larger bathochromic shift was consistent with the narrower HOMO–SOMO and HOMO–LUMO gaps in larger [n]CPP•+ and [n]CPP2+, respectively. In [n]CPP•+, the spins and charges were equally and fully delocalized over the p-phenylene rings of the CPPs, as noted by ESR. 1H NMR revealed that the hydrogen of [n]CPP2+ shifted to a high magnetic field from the neutral compounds due to the diamagnetic ring current derived from the in-plane aromaticity of [n]CPP2+. The single resonances observed in all [n]CPP2+ strongly suggest the complete delocalization of the charges over the CPPs. Furthermore, the contribution of biradical character was clarified for [10]- and [12]CPP by VT-NMR experiment and theoretical calculation.
Co-reporter:Yusuke Sugihara, Shigeru Yamago, and Per B. Zetterlund
Macromolecules 2015 Volume 48(Issue 13) pp:4312-4318
Publication Date(Web):June 17, 2015
DOI:10.1021/acs.macromol.5b00995
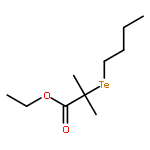
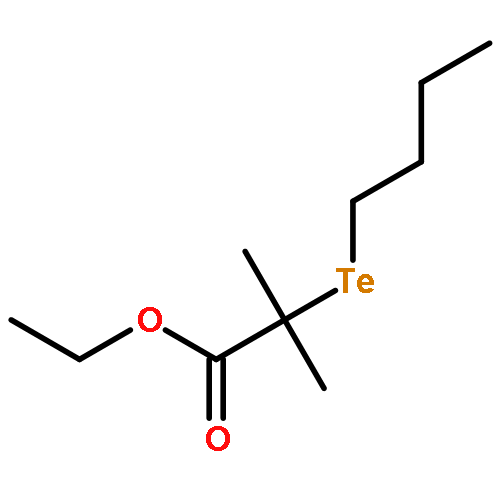
A novel method has been developed for organotellurium-mediated radical polymerization (TERP) in emulsion, herein implemented for styrene using the low molecular weight TERP agent ethyl-2-butyltellanyl-2-methyl propionate (BTEE) at 90 °C in the absence of a conventional radical initiator. The method is based on the use of relatively high concentrations of the two surfactants polyoxyethylene(20) oleyl ether (Brij98; nonionic surfactant) and tetradecyltrimethylammonium bromide (TTAB; cationic surfactant) in the presence of toluene. This is a one-pot approach that does not require high energy mixing nor the use of a water-soluble macroTERP agent, but instead relies on a high initial monomer fraction being located in surfactant micelles (importantly, the system is not a microemulsion). Good control/livingness was obtained in conjunction with satisfactory colloidal stability and relatively narrow particle size distributions with particle diameters mainly in the range well below 100 nm.
Co-reporter:Yasuyuki Nakamura and Shigeru Yamago
Macromolecules 2015 Volume 48(Issue 18) pp:6450-6456
Publication Date(Web):September 4, 2015
DOI:10.1021/acs.macromol.5b01532
A novel method to determine the termination mechanism of radical polymerization, i.e., the selectivity between disproportionation (Disp) and combination (Comb), is developed. The method relies on product analyses of the reaction of polymer-end radicals, which are generated from structurally well-controlled living polymers, and the analyses of molecular weight and end-group structure of the product polymers by GPC, mass spectroscopy, and 1H NMR unambiguously determined the contribution of two competing pathways. The termination mechanism in the polymerization of methyl methacrylate (MMA) and styrene was investigated as a proof of principle of the method by using the corresponding polymers prepared by organotellurium-mediated radical polymerization. The ratios of Disp and Comb (D/C) of poly(methyl methacrylate) (PMMA) or polystyrene (PSt) end radicals at 25 °C were 73/27 or 15/85, respectively, and the results agreed well with the previous reports. The contribution of the Comb increased at higher temperature in both cases, though the temperature dependence was less pronounced in PSt radicals (D/C = 67/37 and 13/87 at 100 °C for PMMA and PSt, respectively). Thermodynamic parameters were determined as ΔΔG‡d/c = (−6.9 ± 0.3) – T × (−14.4 ± 1.0) × 10–3 (kJ mol–1) for PMMA and ΔΔG‡d/c = (−2.0 ± 0.5) – T × (−20.8 ± 1.5) × 10–3 (kJ mol–1) for PSt, in which ΔΔG‡d/c and T are difference in Gibbs energy undergoing Disp and Comb, and temperature in Kelvin, respectively, by carrying out the same experiments between −20 to +100 °C. The parameters reveal that Comb is enthalpically less favored but entropically more favored than Disp in both cases. The effects of molecular weight (chain length) were also investigated, and the D/C ratio became constant when the molecular weight of polymers was more than about 3000.
Co-reporter:Dr. Vijay Kumar Patel;Dr. Eiichi Kayahara;Dr. Shigeru Yamago
Chemistry - A European Journal 2015 Volume 21( Issue 15) pp:5742-5749
Publication Date(Web):
DOI:10.1002/chem.201406650
Abstract
Cyclic precursors of cycloparaphenylenes (CPPs) containing 1,4-dihydroxy-2,5-cyclohexadien-1,4-diyl units are prepared by modifying a synthetic method developed by Jasti and co-workers for the synthesis of corresponding 1,4-dimethoxy derivatives. Reductive aromatization of the diyl moieties by SnCl2/2 HCl takes place under mild conditions and affords the CPPs in good yields, incorporating 5 or 7–12 phenylene units. Highly strained [5]CPP is synthesized in greater than 0.3 g scale. 119Sn NMR spectroscopy clarifies the in situ formation of an ate complex, H2SnCl4, upon mixing a 2:1 ratio of HCl and SnCl2, which serves as a highly active reducing agent under nearly neutral conditions. When more than 2 equivalents of HCl, in relation to SnCl2, are used, acid-catalyzed decomposition of the CPP precursors takes place. The stoichiometry of HCl and SnCl2 is critical in achieving the desired aromatization reaction of highly strained CPP precursors.
Co-reporter:Dr. Vijay Kumar Patel;Dr. Eiichi Kayahara;Dr. Shigeru Yamago
Chemistry - A European Journal 2015 Volume 21( Issue 15) pp:
Publication Date(Web):
DOI:10.1002/chem.201581562
Co-reporter:Takehiro Fujita ; Shigeru Yamago
Chemistry - A European Journal 2015 Volume 21( Issue 51) pp:18547-18550
Publication Date(Web):
DOI:10.1002/chem.201504184
Abstract
Lewis acid (MgBr2)-catalyzed radical polymerization of acrylimides bearing chiral oxazolidinones gave highly isotactic polyacrylimides with up to >99 % meso tetrad (mmm) selectivity. Polymerization in the absence of Lewis acid gave atactic polymers with 80 % racemo diad (r) selectivity; the selectivity was deliberately tuned from 80 % r to >99 % mmm by varying the polymerization conditions. The polyacrylimide was quantitatively converted to corresponding polyacrylates while preserving the stereoregularity, thus providing a general method for the synthesis of atactic to isotactic polyacrylates.
Co-reporter:Dr. Eiichi Kayahara;Rui Qu;Mitsuru Kojima;Dr. Takahiro Iwamoto;Dr. Toshiyasu Suzuki;Dr. Shigeru Yamago
Chemistry - A European Journal 2015 Volume 21( Issue 52) pp:18939-18943
Publication Date(Web):
DOI:10.1002/chem.201504369
Abstract
The syntheses of [3]- and [4]cyclo-9,9-dimethyl-2,7-fluorenes ([3] and [4]CFRs), cyclic trimer, and tetramers of 9,9-dimethyl-2,7-fluorene (FR), respectively, were achieved by the platinum-mediated assembly of FR units and subsequent reductive elimination of platinum. A triangle-shaped tris-platinum complex and a square-shaped tetra-platinum complex were obtained by changing the platinum ligand. The structure of the triangle complex was unambiguously determined by X-ray crystallographic analysis. Reductive elimination of each complex gave [3] and [4]CFRs. Two rotamers of [3]CFR were sufficiently stable at room temperature and were separated by chromatography. The physical properties of the CFRs were also investigated theoretically and experimentally.
Co-reporter:Eiichi Kayahara ; Vijay Kumar Patel
Journal of the American Chemical Society 2014 Volume 136(Issue 6) pp:2284-2287
Publication Date(Web):January 27, 2014
DOI:10.1021/ja413214q
The synthesis of highly strained [5]cycloparaphenylene ([5]CPP), a structural unit of the periphery of C60 and the shortest possible structural constituent of the sidewall of a (5,5) carbon nanotube, was achieved in nine steps in 17% overall yield. The synthesis relied on metal-mediated ring closure of a triethylsilyl (TES)-protected masked precursor 1c followed by removal of the TES groups and subsequent reductive aromatization. UV–vis and electrochemical studies revealed that the HOMO–LUMO gap of [5]CPP is narrow and is comparable to that of C60, as predicted by theoretical calculations. The results suggest that [5]CPP should be an excellent lead compound for molecular electronics.
Co-reporter:Naoyuki Toriumi; Atsuya Muranaka; Eiichi Kayahara; Shigeru Yamago;Masanobu Uchiyama
Journal of the American Chemical Society 2014 Volume 137(Issue 1) pp:82-85
Publication Date(Web):December 19, 2014
DOI:10.1021/ja511320f
The electronic structures of [8]cycloparaphenylene dication ([8]CPP2+) and radical cation ([8]CPP•+) have been investigated by magnetic circular dichroism (MCD) spectroscopy, which enabled unambiguous discrimination between previously conflicting assignments of the UV–vis–NIR absorption spectral bands. Molecular orbital and nucleus-independent chemical shift (NICS) analysis revealed that [8]CPP2+ shows in-plane aromaticity with a (4n + 2) π-electron system (n = 7). This aromaticity appears to be the origin of the unusual stability of the dication. Theoretical calculations further suggested that not only [8]CPP2+ but also all [n]CPP (n = 5–10) dications and dianions exhibit in-plane aromaticity.
Co-reporter:Anthony Kermagoret, Yasuyuki Nakamura, Maxime Bourguignon, Christophe Detrembleur, Christine Jérôme, Shigeru Yamago, and Antoine Debuigne
ACS Macro Letters 2014 Volume 3(Issue 1) pp:114
Publication Date(Web):January 6, 2014
DOI:10.1021/mz400635h
Cobalt-mediated radical polymerization (CMRP) and tellurium-mediated radical polymerization (TERP) were combined for the first time, offering new perspectives in the precision design of macromolecular structures. In particular, the present work highlights the benefits of this strategy for the synthesis of novel poly(vinyl acetate)-based block copolymers. A range of well-defined poly(vinyl acetate)s (PVAc) were first produced via CMRP using the bis(acetylacetonato)cobalt(II) complex (Co(acac)2) as a regulating agent. Substitution of a methyltellanyl moiety for Co(acac)2 at the ω-chain end of the precursor was then achieved upon treatment with dimethylditelluride. In contrast to the PVAc prepared by TERP, the ones produced by sequential CMRP and Co/Te exchange reaction almost exclusively consist of regular head-to-tail-TeMe chain-end species that can be activated by TERP. Ultimately, a series of monomers problematic in Co(acac)2-mediated radical polymerization including N-isopropylacrylamide (NIPAM), 2-(dimethylamino)ethyl acrylate (ADAME), n-butyl acrylate (BA), isoprene (IP), and vinylimidazole (NVIm) were polymerized by TERP from the PVAc–TeMe macroinitiators leading to novel diblock copolymers that cannot be made by each technique used separately.
Co-reporter:Yasuyuki Nakamura;Kouji Nakanishi;Yoshinobu Tsujii;Kenichi Takahashi;Takashi Morinaga;Takaya Sato
Macromolecular Rapid Communications 2014 Volume 35( Issue 6) pp:642-648
Publication Date(Web):
DOI:10.1002/marc.201300855
Co-reporter:Takahiro Iwamoto;Dr. Eiichi Kayahara;Dr. Nobuhiro Yasuda;Dr. Toshiyasu Suzuki;Dr. Shigeru Yamago
Angewandte Chemie International Edition 2014 Volume 53( Issue 25) pp:6430-6434
Publication Date(Web):
DOI:10.1002/anie.201403624
Abstract
A cyclic tetramer of pyrene, [4]cyclo-2,7-pyrenylene ([4]CPY), was synthesized from pyrene in six steps and 18 % overall yield by the platinum-mediated assembly of pyrene units and subsequent reductive elimination of platinum. The structures of the two key intermediates were unambiguously determined by X-ray crystallographic analysis. DFT calculations showed that the topology of the frontier orbitals in [4]CPY was essentially the same as those in [8]cycloparaphenylene ([8]CPP), and that all the pyrene units were fully conjugated. The electrochemical analyses proved the electronic properties of [4]CPY to be similar to those of [8]CPP. The results are in sharp contrast to those obtained for the corresponding linear oligomers of pyrene in which each pyrene unit was electronically isolated. The results clearly show a novel effect of the cyclic structure on cyclic π-conjugated molecules.
Co-reporter:Takahiro Iwamoto;Dr. Zdenek Slanina;Dr. Naomi Mizorogi;Dr. Jingdong Guo;Dr. Takeshi Akasaka;Shigeru Nagase;Dr. Hikaru Takaya;Dr. Nobuhiro Yasuda;Dr. Tatsuhisa Kato;Dr. Shigeru Yamago
Chemistry - A European Journal 2014 Volume 20( Issue 44) pp:14403-14409
Publication Date(Web):
DOI:10.1002/chem.201403879
Abstract
[11]Cycloparaphenylene ([11]CPP) selectively encapsulates La@C82 to form the shortest possible metallofullerene–carbon nanotube (CNT) peapod, La@C82⊂[11]CPP, in solution and in the solid state. Complexation in solution was affected by the polarity of the solvent and was 16 times stronger in the polar solvent nitrobenzene than in the nonpolar solvent 1,2-dichlorobenzene. Electrochemical analysis revealed that the redox potentials of La@C82 were negatively shifted upon complexation from free La@C82. Furthermore, the shifts in the redox potentials increased with polarity of the solvent. These results are consistent with formation of a polar complex, (La@C82)δ−⊂[11]CPPδ+, by partial electron transfer from [11]CPP to La@C82. This is the first observation of such an electronic interaction between a fullerene pea and CPP pod. Theoretical calculations also supported partial charge transfer (0.07) from [11]CPP to La@C82. The structure of the complex was unambiguously determined by X-ray crystallographic analysis, which showed the La atom inside the C82 near the periphery of the [11]CPP. The dipole moment of La@C82 was projected toward the CPP pea, nearly perpendicular to the CPP axis. The position of the La atom and the direction of the dipole moment in La@C82⊂[11]CPP were significantly different from those observed in La@C82⊂CNT, thus indicating a difference in orientation of the fullerene peas between fullerene–CPP and fullerene–CNT peapods. These results highlight the importance of pea–pea interactions in determining the orientation of the metallofullerene in metallofullerene–CNT peapods.
Co-reporter:Takahiro Iwamoto;Dr. Eiichi Kayahara;Dr. Nobuhiro Yasuda;Dr. Toshiyasu Suzuki;Dr. Shigeru Yamago
Angewandte Chemie 2014 Volume 126( Issue 25) pp:6548-6552
Publication Date(Web):
DOI:10.1002/ange.201403624
Abstract
A cyclic tetramer of pyrene, [4]cyclo-2,7-pyrenylene ([4]CPY), was synthesized from pyrene in six steps and 18 % overall yield by the platinum-mediated assembly of pyrene units and subsequent reductive elimination of platinum. The structures of the two key intermediates were unambiguously determined by X-ray crystallographic analysis. DFT calculations showed that the topology of the frontier orbitals in [4]CPY was essentially the same as those in [8]cycloparaphenylene ([8]CPP), and that all the pyrene units were fully conjugated. The electrochemical analyses proved the electronic properties of [4]CPY to be similar to those of [8]CPP. The results are in sharp contrast to those obtained for the corresponding linear oligomers of pyrene in which each pyrene unit was electronically isolated. The results clearly show a novel effect of the cyclic structure on cyclic π-conjugated molecules.
Co-reporter:Yasuyuki Nakamura, Takahiro Arima, and Shigeru Yamago
Macromolecules 2014 Volume 47(Issue 2) pp:582-588
Publication Date(Web):January 9, 2014
DOI:10.1021/ma402354m
Photoirradiation of structurally well-defined “living” polymers prepared by organotellurium-mediated living radical polymerization in the presence of dienes or styrenes induced selective polymer-end coupling reaction with the concomitant insertion of the dienes or styrenes with >90% coupling efficiency. The number of inserted dienes or styrenes could be highly controlled to two molecules when acrylic polymers were used. Therefore, various mid-chain-functionalized polymers with well-controlled molecular and macromolecular structure in terms of their molecular weight, molecular weight distribution, functionality, and position were successfully synthesized by employing functionalized dienes or styrenes. The method was applied to the facile synthesis of mid-chain-functionalized telechelic polymers and a 4-miktoarm star polymer with a well-controlled structure.
Co-reporter:Shigeru Yamago;Eiichi Kayahara;Takahiro Iwamoto
The Chemical Record 2014 Volume 14( Issue 1) pp:84-100
Publication Date(Web):
DOI:10.1002/tcr.201300035
Abstract
This article describes the most recent developments in the synthesis of three-dimensional π-conjugated molecules and the elucidation of their properties made by our research group. Various cycloparaphenylenes (CPPs) of different sizes and a cage-like 3D molecule were synthesized based on the platinum-mediated assembly of π-units and subsequent reductive elimination of platinum. The assembly of π-units by this method mimics the self-assembly process for the formation of supramolecular ligand–metal complexes with 3D cages and polyhedral structures. Furthermore, reductive elimination of platinum successfully took place with high efficiency, despite the high strain energy of the target molecule. Several size-dependent physical properties of CPPs, namely the photophysical, redox, and host–guest chemistries, were also clarified. These results are of use for a molecular-level understanding of CNT physical properties as well as fullerene peapods. Theoretical and electrochemical studies suggest that small CPPs and their derivatives should be excellent lead compounds for molecular electronics.
Co-reporter:Shigeru Yamago, Yasuyuki Nakamura
Polymer 2013 Volume 54(Issue 3) pp:981-994
Publication Date(Web):5 February 2013
DOI:10.1016/j.polymer.2012.11.046
The effects of photoirradiation in controlled and living radical polymerization (LRP), namely nitroxide-mediated polymerization (NMP), atom-transfer radical polymerization (ATRP), cobalt-mediated radical polymerization (CMRP), reversible addition-fragmentation chain transfer polymerization (RAFT), organoiodine-mediated radical polymerization (IRP), and organotellurium-mediated radical polymerization (TERP), are summarized. As in the conventional radical polymerization, photoirradiation has been used for generating radicals under mild conditions in LRP methods. In addition to this use, photoirradiation is also used to overcome the difficulties characteristic to each method, such as activation of catalysis, generation of controlling agents, and increasing the polymer-end structure. The most-recent developments in the use of photochemistry in LRP are summarized in this review.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Dr. Eiichi Kayahara;Takahiko Kouyama;Dr. Tatsuhisa Kato;Dr. Hikaru Takaya;Dr. Nobuhiro Yasuda;Dr. Shigeru Yamago
Angewandte Chemie 2013 Volume 125( Issue 51) pp:13967-13971
Publication Date(Web):
DOI:10.1002/ange.201306881
Co-reporter:Takahiro Iwamoto;Yoshiki Watanabe;Dr. Hikaru Takaya;Dr. Takeharu Haino;Dr. Nobuhiro Yasuda;Dr. Shigeru Yamago
Chemistry - A European Journal 2013 Volume 19( Issue 42) pp:14061-14068
Publication Date(Web):
DOI:10.1002/chem.201302694
Abstract
The size- and orientation-selective formation of the shortest-possible C70 peapod in solution and in the solid state by using the shortest structural unit of an “armchair” carbon nanotube (CNT), cycloparaphenylene (CPP), has been studied. [10]CPP and [11]CPP exothermically formed 1:1 complexes with C70, thereby giving the resulting peapods. A van′t Hoff plot analysis revealed that the formation of these complexes in 1,2-dichlorobenzene was mainly driven by entropy, whereas the theoretical calculations suggested that the formation of the complex in the gas phase was predominantly driven by enthalpy. C70 was found to exist in two distinct orientations inside the CPP cavity, namely “lying” and “standing”, depending on the specific size of the CPP. The theoretical calculations and the X-ray crystallographic analysis revealed that the interactions between [10]CPP and the short axis of C70 in its lying orientation were isotropic and similar to those observed between [10]CPP and C60. However, the interactions between [11]CPP and C70 in its standing orientation were anisotropic, thereby involving the radial deformation of [11]CPP into an ellipsoidal shape. This “induced fit” maximized the van der Waals interactions with the long axis of C70. Theoretical calculations revealed that the deformation occurred readily with low energy loss, thus suggesting that CPPs are highly radially elastic molecules. These results also indicate that the same type of radial deformation should occur in CNT peapods that encapsulate anisotropic fullerenes.
Co-reporter:Takahiro Iwamoto;Yoshiki Watanabe;Dr. Hikaru Takaya;Dr. Takeharu Haino;Dr. Nobuhiro Yasuda;Dr. Shigeru Yamago
Chemistry - A European Journal 2013 Volume 19( Issue 42) pp:
Publication Date(Web):
DOI:10.1002/chem.201390168
Co-reporter:Dr. Eiichi Kayahara;Takahiko Kouyama;Dr. Tatsuhisa Kato;Dr. Hikaru Takaya;Dr. Nobuhiro Yasuda;Dr. Shigeru Yamago
Angewandte Chemie International Edition 2013 Volume 52( Issue 51) pp:13722-13726
Publication Date(Web):
DOI:10.1002/anie.201306881
Co-reporter:Yasuyuki Nakamura ; Takahiro Arima ; Sora Tomita
Journal of the American Chemical Society 2012 Volume 134(Issue 12) pp:5536-5539
Publication Date(Web):March 13, 2012
DOI:10.1021/ja300869x
An efficient and simple method for the synthesis of symmetric macromolecules by photoinduced switching from radical polymerization to a radical coupling reaction is reported. Structurally well-defined telechelic polyisoprenes and ABA-triblock copolymers were prepared by successive organotellurium-mediated living radical polymerization (TERP) under thermal conditions, followed by a polymer-end radical coupling reaction under photoirradiation.
Co-reporter:Eiichi Kayahara, Yoichi Sakamoto, Toshiyasu Suzuki, and Shigeru Yamago
Organic Letters 2012 Volume 14(Issue 13) pp:3284-3287
Publication Date(Web):June 18, 2012
DOI:10.1021/ol301242t
[10]Cycloparaphenylene ([10]CPP) was selectively synthesized in four steps in 13% overall yield from commercially available 4,4′-diiodobiphenyl by using mono-I–Sn exchange, Sn–Pt transmetalation, I–Pd exchange, and subsequent oxidative coupling reactions. The single-crystal X-ray structure of [10]CPP is described.
Co-reporter:Shuo Feng, Weijian Xu, Koji Nakanishi, and Shigeru Yamago
ACS Macro Letters 2012 Volume 1(Issue 1) pp:146
Publication Date(Web):December 8, 2011
DOI:10.1021/mz200133d
Ionic liquids (ILs) served perfectly as solvents for the organotellurium-mediated living radical polymerization (TERP) of methyl methacrylate (MMA), methyl acrylate (MA), and styrene. The reaction rate of polymerizing MMA and MA was significantly increased as previously reported, and the controllability of the polydispersity index (PDI) was also improved by a great margin. The TERP of MMA can now give poly(methyl methacrylates) (PMMAs) with PDIs less than 1.1 and nearly full conversion in a half hour without the presence of (TeMe)2. The kinetic study revealed that the improved control could be ascribed to a faster degenerative chain transfer (DT) reaction which plays a key role in the control of PDI for TERP. Besides the polar effect of ILs, the existence of Lewis acid–base interaction between ILs and the Te atom was proven by UV–vis spectroscopy and 125Te NMR results. Such positive interaction lowered the activation energy of the DT process.
Co-reporter:Eri Mishima
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 11) pp:2254-2264
Publication Date(Web):
DOI:10.1002/pola.26004
Abstract
Random and alternating copolymerizations of acrylates, methacrylates, acrylonitorile, and acrylamides with vinyl ethers under organotellurium-, organostibine-, and organobismuthine-mediated living radical polymerization (TERP, SBRP, and BIRP, respectively) have been studied. Structurally well-controlled random and alternating copolymers with controlled molecular weights and polydispersities were synthesized. The highly alternating copolymerization occurred in a combination of acrylates and vinyl ethers and acrylonitorile and vinyl ethers by using excess amount of vinyl ethers over acrylates and acrylonitorile. On the contrary, alternating copolymerization did not occur in a combination of acrylamides and vinyl ethers even excess amount of vinyl ethers were used. The reactivity of polymer-end radicals to a vinyl ether was estimated by the theoretical calculations, and it was suggested that the energy level of singly occupied molecular orbital (SOMO) of polymer-end radical species determined the reactivity. By combining living random and alternating copolymerization with living radical or living cationic polymerization, new block copolymers, such as (PBA-alt-PIBVE)-block-(PtBA-co-PIBVE), PBA-block-(PBA-alt-PIBVE), and (PTFEA-alt-PIBVE)-block-PIBVE, with controlled macromolecular structures were successfully synthesized. © 2012 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Eri Mishima, Tomoki Tamura, and Shigeru Yamago
Macromolecules 2012 Volume 45(Issue 7) pp:2989-2994
Publication Date(Web):April 2, 2012
DOI:10.1021/ma300325r
Copolymerization of acrylates, such as methyl acrylates (MAs) and trifluoroethyl acrylates (TFEAs), and 6-methyleneundecane (6MU), under organotellurium-mediated living radical polymerization (TERP) conditions, was investigated. Structurally well-controlled copolymers, poly(MA-co-6MU) and poly(TFEA-co-6MU), with predetermined number-average molecular weights (Mn = 3000–14000) and low polydispersity indices (PDI = 1.14 to 1.45) were obtained under thermal condition in the presence of an azo-initiator or photoirradiation. The addition of protic acids, such as 1,3-C6H4[C(CF3)2OH]2 and (CF3)2CHOH, was effective to increase the insertion of 6MU into the copolymer. The structure of the ω-polymer-end group was analyzed by using labeling experiments, which revealed that the majority of the end groups were derived from 6MU. Although activation of dormant species derived from α-olefin is difficult, the copolymers were successfully reactivated and served as macro-chain-transfer agents for the synthesis of block copolymers. Poly(MA-co-6MU)-block-styrene and poly(MA-co-6MU)-block-(N-vinylpyrrolidone) with controlled structures were synthesized for the first time.
Co-reporter:Eri Mishima, Tomoki Tamura, and Shigeru Yamago
Macromolecules 2012 Volume 45(Issue 22) pp:8998-9003
Publication Date(Web):November 7, 2012
DOI:10.1021/ma301570r
Copolymerization of 1-octene and (meth)acrylates, such as methyl acrylate, trifluoroethyl acrylate (TFEA), methyl methacrylate, and trifluoroethyl methacrylate, under organotellurium-mediated living radical polymerization (TERP) conditions was investigated. Polymerization under thermal conditions gave copolymers with considerably broad molecular distributions (polydispersity index [PDI] > 1.45), whereas that under photoirradiation greatly increased the PDI control. Structurally well-controlled copolymers with number-average molecular weights (Mn) of 3000–18 000 and low PDIs (1.22–1.45) were obtained. Addition of Brønsted acids, such as 1,3-C6H4[C(CF3)2OH]2 and hexafluoroisopropanol, increased the insertion of 1-octene into the copolymer. The molar fraction of 1-octene (MFoct) reached ∼0.5 in the copolymerization using TFEA as an acrylate monomer and excess amount of 1-octene in the presence of the acid. The copolymer was used as a macro-chain-transfer agent for the synthesis of block copolymers. This is the first example of the use of this type of copolymer as a macro-chain-transfer agent in the controlled synthesis of block copolymers.
Co-reporter:Eri Mishima
Macromolecular Rapid Communications 2011 Volume 32( Issue 12) pp:893-898
Publication Date(Web):
DOI:10.1002/marc.201100089
Co-reporter:Eri Mishima
Macromolecular Rapid Communications 2011 Volume 32( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/marc.201190027
Co-reporter:Eiichi Kayahara;Noriaki Kondo
Heteroatom Chemistry 2011 Volume 22( Issue 3-4) pp:307-315
Publication Date(Web):
DOI:10.1002/hc.20681
Abstract
The substituent effect on the antimony atom in organostibine-mediated degenerative transfer living radical polymerization has been studied. 2-Diphenylstibanyl-2-methylpropionitrile (1a) was synthesized as an organostibine chain transfer agent (CTA), and its effect was compared with the previously reported dimethylstibanyl analogue 1b. Both CTAs successfully polymerized butyl acrylate (BA) in a highly controlled manner giving poly(butyl acrylate)s possessing number average molecular weights close to the theoretical values determined by BA/1 ratios and narrow molecular weight distribution. The controllability of styrene polymerization by 1a was slightly decreased but the decrease is still at an acceptable level. However, 1a did not control the polymerization of methyl methacrylate (MMA). The results are in sharp contrast that 1b can highly control the polymerization of styrene and MMA. Kinetic studies using diphenylstibanyl-substituted polystyrene macro-CTA 2 in styrene polymerization revealed that the degenerative chain transfer reaction of the diphenylstibanyl group takes place faster than that of the dimethylstibanyl group, suggesting that the reactivity of the diphenylstibanyl group is not the origin of the low control. Chain extension experiments revealed the formation of a considerable amount of dead polymers when 1a was used as a CTA, and the frequent occurrence of termination reaction is the most probable origin of the low control. The addition of tetraphenyldistibine significantly increased the control of styrene and MMA polymerization, and structurally well-defined polystyrene and poly(methyl methacrylate) were obtained by applying a binary system consisting from 1a and tetraphenyldistibine. © 2011 Wiley Periodicals, Inc. Heteroatom Chem 22:307–315, 2011; View this article online at wileyonlinelibrary.com. DOI 10.1002/hc.20681
Co-reporter:Takahiro Iwamoto;Yoshiki Watanabe;Tatsuya Sadahiro; Takeharu Haino; Shigeru Yamago
Angewandte Chemie 2011 Volume 123( Issue 36) pp:8492-8494
Publication Date(Web):
DOI:10.1002/ange.201102302
Co-reporter:Takahiro Iwamoto;Yoshiki Watanabe;Tatsuya Sadahiro; Takeharu Haino; Shigeru Yamago
Angewandte Chemie International Edition 2011 Volume 50( Issue 36) pp:8342-8344
Publication Date(Web):
DOI:10.1002/anie.201102302
Co-reporter:Yasuyuki Nakamura, Yukie Kitada, Yu Kobayashi, Biswajit Ray, and Shigeru Yamago
Macromolecules 2011 Volume 44(Issue 21) pp:8388-8397
Publication Date(Web):October 13, 2011
DOI:10.1021/ma201761q
The effects of azo initiators on the structures of the α-polymer chain ends in organotellurium-, organostibine-, organobismuthine-, and organoiodine-mediated living radical polymerizations (TERP, SBRP, BIRP, and IRP, respectively) and reversible addition–fragmentation chain transfer radical polymerization (RAFT) were quantitatively analyzed for the first time. These polymerization methods predominantly, or exclusively, follow the degenerative chain transfer (DT) mechanism for the activation and deactivation of dormant and radical species. An external radical species is required to initiate the polymerization under DT conditions, and the influx of radical species decreases the α-end-group fidelity. The effect was examined for polymerizations of styrene (St), n-butyl acrylate (BA), N-vinylpyrrolidone (NVP), methyl methacrylate (MMA), and isoprene (Ip) in the presence of different chain transfer agents (CTAs) and azo initiators, such as 2,2′-azobis(isobutyronitrile) (AIBN), 1,1′-azobis(cyclohexane-1-carbonitrile) (ACHN), and 1-[(1-cyano-1-methylethyl)azo]formamide (V-30), and the formed polymers 2 and 3, which possess α-end structures derived from the CTA and azo initiator, respectively, were analyzed by using matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF MS). The amount of 3 was negligible when the rate of propagation (kp) was sufficiently high, such as in the polymerizations of BA, MMA, and NVP. The formation of 3 was considerable when kp was low, such as in the polymerizations of St and Ip, but was considerably reduced by carrying out the polymerization at high temperatures using an azo initiator decomposed at high temperatures. Furthermore, the formation of 3 was completely disappeared when the initiating radical was supplied by the reversible termination (RT) mechanism in TERP. The results indicate that α-polymer chain end structure is better controlled when both RT and DT are involved. The effect of azo initiator on the synthesis of a block copolymer was also examined, and pure diblock copolymer PMMA-b-PSt was obtained when both RT and DT were involved.
Co-reporter:Eiichi Kayahara;Hiroto Yamada;Dr. Shigeru Yamago
Chemistry - A European Journal 2011 Volume 17( Issue 19) pp:5272-5280
Publication Date(Web):
DOI:10.1002/chem.201100265
Abstract
Generation of carbanions from organostibines and organobismuthines through heteroatom–metal exchange reactions was examined from synthetic and mechanistic viewpoints. The exchange reaction proceeded spontaneously upon treatment with various organometallic reagents, such as alkyl lithiums, tetraalkyl zincates, and alkyl magnesium halides to afford the corresponding carbanions quantitatively. Due to the high reactivity of these heteroatom compounds, the exchange reactions took place exclusively even in the presence of various polar functional groups, which potentially react with organometallic species. The advantage of this method was exemplified by the end-group transformation of living polymers that bear these heteroatom species at the ω-polymer end, prepared by using organostibine and bismuthine-mediated living radical polymerizations. Various polymers that bear polar functional groups and acidic hydrogen—for example, poly(methyl methacrylate), poly(butyl acrylate), poly(N-isopropyl acrylamide), and poly(2-hydroxyethyl methacrylate)—could be used in the exchange reactions, and subsequent trapping with electrophiles afforded the corresponding polymers with controlled molecular weights, molecular weight distributions, and end-group functionalities. Competition experiments showed that organostibines and organobismuthines were among the most reactive heteroatom compounds towards organometallic reagents and that their high reactivity was responsible for the high chemoselectivity in the exchange reaction.
Co-reporter:Eiichi Kayahara;Hiroto Yamada;Dr. Shigeru Yamago
Chemistry - A European Journal 2011 Volume 17( Issue 19) pp:
Publication Date(Web):
DOI:10.1002/chem.201190092
Co-reporter:Shigeru Yamago ;Yoshiki Watanabe ;Takahiro Iwamoto
Angewandte Chemie International Edition 2010 Volume 49( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/anie.200906851
Co-reporter:Shigeru Yamago ;Yoshiki Watanabe ;Takahiro Iwamoto
Angewandte Chemie International Edition 2010 Volume 49( Issue 4) pp:757-759
Publication Date(Web):
DOI:10.1002/anie.200905659
Co-reporter:Shigeru Yamago ;Yoshiki Watanabe ;Takahiro Iwamoto
Angewandte Chemie 2010 Volume 122( Issue 4) pp:769-771
Publication Date(Web):
DOI:10.1002/ange.200905659
Co-reporter:Shigeru Yamago ;Yoshiki Watanabe ;Takahiro Iwamoto
Angewandte Chemie 2010 Volume 122( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/ange.200906851
Co-reporter:Shigeru Yamago
Chemical Reviews 2009 Volume 109(Issue 11) pp:5051
Publication Date(Web):August 28, 2009
DOI:10.1021/cr9001269
Co-reporter:Shigeru Yamago, Eiichi Kayahara, Hiroto Yamada
Reactive and Functional Polymers 2009 69(7) pp: 416-423
Publication Date(Web):July 2009
DOI:10.1016/j.reactfunctpolym.2009.03.008
Co-reporter:Shigeru Yamago Dr.;Takeshi Yamada Dr.;Manabu Togai;Yuu Ukai;Eiichi Kayahara;Na Pan Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 4) pp:1018-1029
Publication Date(Web):
DOI:10.1002/chem.200801754
Abstract
Several organostibine chain-transfer agents possessing polar functional groups have been prepared by the reactions of azo initiators and tetramethyldistibine (1). Carbon-centered radicals thermally generated from the azo initiators were trapped by 1 to yield the corresponding organostibine chain-transfer agents. The high yields observed in the synthesis of the chain-transfer agents strongly suggest that distibines have excellent radicophilic reactivity. As the reactions proceeded under neutral conditions, functional groups that are incompatible with ionic conditions were incorporated into the chain-transfer agents. The chain-transfer agents were used in living radical polymerization to synthesize the corresponding α-functionalized polymers. As the functional groups in the chain-transfer agents did not interfere with the polymerization reaction, well-controlled polymers possessing number-average molecular weights (Mns) predetermined by the monomer/transfer agent ratios were synthesized with low polydispersity indices (PDIs). The organostibanyl ω-polymer ends were transformed into a number of different functional groups by radical-coupling, radical-addition, and oxidation reactions. Therefore, it was possible to synthesize well-controlled telechelic polymers with the same and also with different functional groups at their α- and ω-polymer ends. Distibine 1 was also found to increase PDI control in the living radical polymerization of styrene and methyl methacrylate (MMA) using a purified organostibine chain-transfer agent. Well-controlled poly(methyl methacrylate)s with Mn values ranging from 10 000 to 120 000 with low PDIs (1.05–1.15) were synthesized by the addition of a catalytic amount of 1. The results have been attributed to the high reactivity of distibine 1 towards polymer-end radicals, which are spontaneously deactivated to yield organostibine dormant species.
Co-reporter:Shigeru Yamago Dr.;Eiichi Kayahara;Masashi Kotani Dr.;Biswajit Ray Dr.;Yungwan Kwak Dr.;Atsushi Goto Dr.;Takeshi Fukuda Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 8) pp:
Publication Date(Web):5 JAN 2007
DOI:10.1002/anie.200604473
The living end: Organobismuthines promote highly controlled living radical polymerization through two activation mechanisms, namely, thermal generation and degenerative transfer (see scheme). Both conjugated and nonconjugated vinyl monomers are polymerized to give well-defined polymers with predetermined molecular weight (Mn) and low polydispersity index (PDI).
Co-reporter:Shigeru Yamago Dr.;Eiichi Kayahara;Masashi Kotani Dr.;Biswajit Ray Dr.;Yungwan Kwak Dr.;Atsushi Goto Dr.;Takeshi Fukuda Dr.
Angewandte Chemie 2007 Volume 119(Issue 8) pp:
Publication Date(Web):5 JAN 2007
DOI:10.1002/ange.200604473
Lebendiges Ende: Organobismutine katalysieren eine kontrollierte lebende radikalische Polymerisation über zwei Aktivierungsmechanismen: thermische Erzeugung und degenerativer Transfer (siehe Schema). Sowohl konjugierte als auch nichtkonjugierte Vinylmonomere werden zu gut definierten Polymeren mit vorherbestimmtem Molekulargewicht (Mn) und niedrigem Polydispersitätsindex (PDI) polymerisiert.
Co-reporter:Takeshi Yamada Dr.;Kazunobu Takemura;Jun-ichi Yoshida
Angewandte Chemie 2006 Volume 118(Issue 45) pp:
Publication Date(Web):20 OCT 2006
DOI:10.1002/ange.200602699
Doppelaufgabe: Ein Phosphorsäureester an der C2-Position von Glycosyldonoren bewirkt eine selektive Glycosylierung am anomeren Kohlenstoffatom mit vollständiger 1,2-trans-Selektivität. Da der Phosphorsäureester unter Freisetzung der Hydroxygruppe gespalten werden kann, fungiert die Gruppe in der Oligosaccharidsynthese als stereodirigierende Schutzgruppe.
Co-reporter:Takeshi Yamada Dr.;Kazunobu Takemura;Jun-ichi Yoshida
Angewandte Chemie International Edition 2006 Volume 45(Issue 45) pp:
Publication Date(Web):20 OCT 2006
DOI:10.1002/anie.200602699
Double duty: A phosphoric ester at the C2 position in glycosyl donors directs the glycosylation to occur selectively at the anomeric carbon atom with complete 1,2-trans selectivity. Since the phosphoric ester can be removed to give the hydroxy function, this group serves as a stereodirecting protecting group in oligosaccharide synthesis.
Co-reporter:Takahiro Iwamoto ; Yoshiki Watanabe ; Youichi Sakamoto ; Toshiyasu Suzuki
Journal of the American Chemical Society () pp:
Publication Date(Web):May 4, 2011
DOI:10.1021/ja2020668
[n]Cycloparaphenylenes (n = 8–13, CPPs) were synthesized, and their physical properties were systematically investigated. [8] and [12]CPPs were selectively prepared from the reaction of 4,4′-bis(trimethylstannyl)biphenyl and 4,4′′-bis(trimethylstannyl)terphenyl, respectively, with Pt(cod)Cl2 (cod = 1,5-cyclooctadiene) through square-shaped tetranuclear platinum intermediates. A mixture of [8]–[13]CPPs was prepared in good combined yields by mixing biphenyl and terphenyl precursors with platinum sources. Products were easily separated and purified by using gel permeation chromatography. In 1H NMR spectra, the proton of the CPPs shifts to a lower field as n increased due to an anisotropic effect from the nearby PP moieties. Although the UV–vis spectra were rather insensitive to the size of the CPPs, the fluorescence spectra changed significantly in relation to their size. A larger Stokes shift was observed for the smaller CPPs. Redox properties of the CPPs were measured for the first time by using cyclic voltammetry, and the smaller CPPs had lower oxidation potentials. The results are consistent with the HOMO energies of CPPs, of which the smaller CPPs had higher energies.
Co-reporter:Shigeru Yamago ; Yoshikazu Yahata ; Kouji Nakanishi ; Shota Konishi ; Eiichi Kayahara ; Akihiro Nomura ; Atsushi Goto ;Yoshinobu Tsujii
Macromolecules () pp:
Publication Date(Web):August 23, 2013
DOI:10.1021/ma401385a
An organotellurium chain transfer agent (CTA) bearing a triethoxysilyl group at one end and a 2-methyltellanyl-2-methylpropionate group at the other was prepared and immobilized on the surface of a silicon wafer and silica nanoparticle (SiP). Surface-initiated organotellurium-mediated living radical polymerization (SI-TERP) from the immobilized CTA in the presence of nonimmobilized (free) organotellurium CTA was examined. Concentrated polymer brushes (CPBs) having surface occupancies above 0.1 were prepared by polymerization of various monomers, including styrene, methyl methacrylate, butyl acrylate, N-isopropyl acrylamide, N-vinyl pyrrolidone (NVP), and N-vinyl carbazole (NVC). All CPBs were formed in a controlled manner, with number-average molecular weights close to the theoretical values and low polydispersity indices (<1.41). Structurally well-controlled CPBs comprising unconjugated monomers, NVP and NVC, were prepared for the first time. Atomic force microscopy and transmission electron microscopy analyses of the CPBs revealed the highly stretched and anisotropic structure of the grafted polymer chain in a good solvent.





![Propanoic acid, 2-methyl-2-[(2,2,6,6-tetramethyl-1-piperidinyl)oxy]-, ethyl ester](/data/chemimg/3764300/212128-91-1.png)
![Propanoic acid, 2-methyl-2-[(2,2,6,6-tetramethyl-1-piperidinyl)oxy]-, ethyl ester](/data/chemimg/3764300/212128-91-1_b.png)


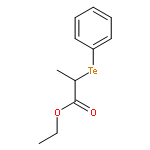
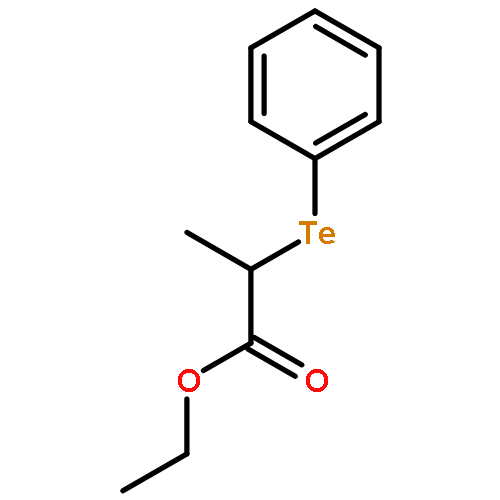
![Benzene, [(1-phenylethyl)telluro]-](http://img.cochemist.com/ccimg/121400/121335-32-8.png)
![Benzene, [(1-phenylethyl)telluro]-](http://img.cochemist.com/ccimg/121400/121335-32-8_b.png)



![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2_b.png)







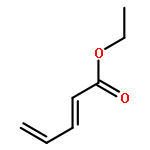
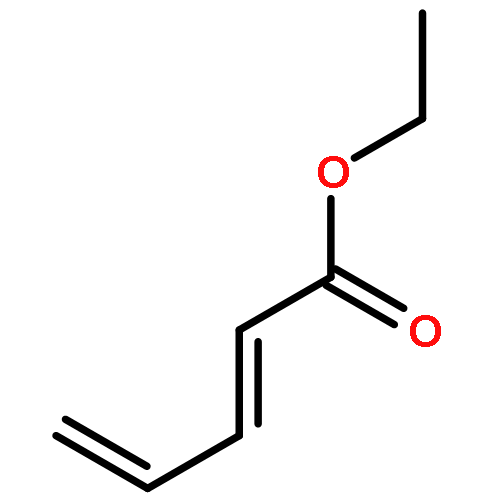
![[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]dichloroplatinum](http://img.cochemist.com/ccimg/12200/12152-26-0.png)
![[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]dichloroplatinum](http://img.cochemist.com/ccimg/12200/12152-26-0_b.png)


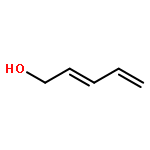
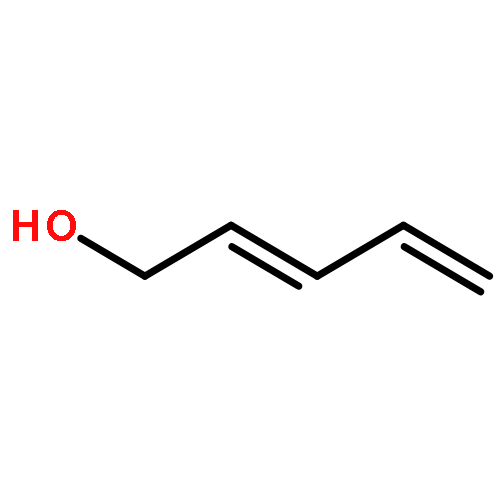


![Decacyclo[32.2.2.22,5.26,9.210,13.214,17.218,21.222,25.226,29.230,33]tetrapentaconta-1,3,5,7,9,11,13,15,17,19,21,23,25,27,29,31,33,35,37,39,41,43,45,47,49,51,53-heptacosaene](/data/chemimg/3742200/1092522-74-1.png)
![Decacyclo[32.2.2.22,5.26,9.210,13.214,17.218,21.222,25.226,29.230,33]tetrapentaconta-1,3,5,7,9,11,13,15,17,19,21,23,25,27,29,31,33,35,37,39,41,43,45,47,49,51,53-heptacosaene](/data/chemimg/3742200/1092522-74-1_b.png)


![Nonacyclo[28.2.2.22,5.26,9.210,13.214,17.218,21.222,25.226,29]octatetraconta-1,3,5,7,9,11,13,15,17,19,21,23,25,27,29,31,33,35,37,39,41,43,45,47-tetracosaene](/data/chemimg/3742600/1217269-85-6.png)
![Nonacyclo[28.2.2.22,5.26,9.210,13.214,17.218,21.222,25.226,29]octatetraconta-1,3,5,7,9,11,13,15,17,19,21,23,25,27,29,31,33,35,37,39,41,43,45,47-tetracosaene](/data/chemimg/3742600/1217269-85-6_b.png)



![Heptacyclo[20.2.2.22,5.26,9.210,13.214,17.218,21]hexatriaconta-1,3,5,7,9,11,13,15,17,19,21,23,25,27,29,31,33,35-octadecaene](/data/chemimg/3742600/156980-13-1.png)
![Heptacyclo[20.2.2.22,5.26,9.210,13.214,17.218,21]hexatriaconta-1,3,5,7,9,11,13,15,17,19,21,23,25,27,29,31,33,35-octadecaene](/data/chemimg/3742600/156980-13-1_b.png)
![[5]Cycloparaphenylene](http://img.cochemist.com/ccimg/96200/96100-94-6.png)
![[5]Cycloparaphenylene](http://img.cochemist.com/ccimg/96200/96100-94-6_b.png)