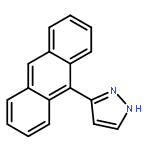
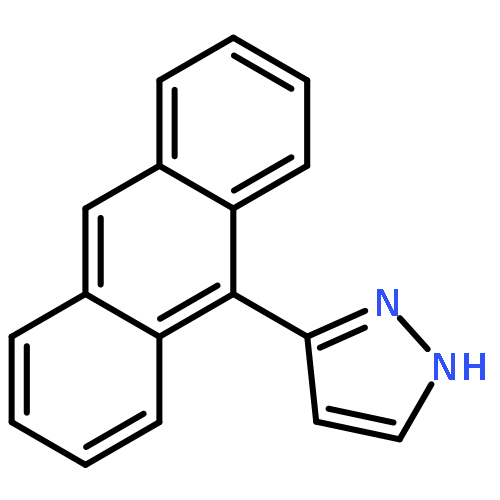
Co-reporter:Hai-Na Zhang;Qing-Fu Zhang;Jia-Jia Wang;Ai-Jing Geng
Acta Crystallographica Section C 2014 Volume 70( Issue 3) pp:292-296
Publication Date(Web):
DOI:10.1107/S205322961400312X
The title compound, [Ag(C15H11N4O2S)]n, was synthesized by the reaction of 4-{[(1-phenyl-1H-tetrazol-5-yl)sulfanyl]methyl}benzoic acid (Hptmba) with silver nitrate and triethylamine at room temperature. The asymmetric unit contains one crystallographically independent AgI cation and one ptmba− ligand. Each AgI cation is tricoordinated by two carboxylate O atoms and one tetrazole N atom from three different ptmba− ligands, displaying a distorted T-shaped geometry. Three AgI cations are linked by tris-monodentate bridging ptmba− ligands to form a one-dimensional double chain along the c axis, which is further consolidated by an intrachain π–π contact with an offset face-to-face distance of 4.176 (3) Å between the centroids of two adjacent aromatic rings in neighbouring benzoate groups. The one-dimensional chains are linked into a three-dimensional supramolecular framework by additional π–π interchain interactions, viz. of 3.753 (3) Å between two phenyl substituents of the tetrazole rings and of 4.326 (2) Å between a benzoate ring and a tetrazole ring. Thermogravimetric analysis and the fluorescence spectrum of the title compound reveal its good thermal stability and a strong green luminescence at room temperature.
Co-reporter:Wei ZHANG, Fan-Jun MENG, Mei-Juan ZHOU, Peng LI, Jing-Jing ZHAO, Huai-Sheng WANG, Ji-Feng LIU, Chong ZHANG
Chinese Journal of Analytical Chemistry 2013 Volume 41(Issue 5) pp:687-692
Publication Date(Web):May 2013
DOI:10.1016/S1872-2040(13)60651-9
Co-reporter:Qingfu Zhang;Haina Zhang;Jinyun Wang;Dezhi Sun
Heteroatom Chemistry 2012 Volume 23( Issue 5) pp:435-443
Publication Date(Web):
DOI:10.1002/hc.21034
Abstract
A new thiotetrazole compound, 4-((1-phenyl-1H-tetrazol-5-ylthio)methyl) benzoic acid (1), has been synthesized and characterized by elemental analysis, 1H and 13C NMR, ESI-MS, FT-IR, UV–vis, fluorescence spectra, and single-crystal X-ray diffraction analysis. The structural analysis reveals that compound 1 adopts a nonplanar geometric structure and exhibits an extensive but not uniform π delocalization with all members of the tetrazolyl ring and the exocyclic sulfur atom. A density functional theory (DFT) calculation at B3LYP/6-31G** level of theory was performed to elucidate the structure of this thiotetrazole system. And the time-dependent DFT (TD-DFT) calculations of absorption spectra reveal two electron-transition bands derived from the contribution of π π* transitions, which are in agreement with experimental results. Moreover, compound 1 exhibits a blue-light emission (λem = 441 nm) in the solid state at room temperature. © 2012 Wiley Periodicals, Inc. Heteroatom Chem 23:435–443, 2012; View this article online at wileyonlinelibrary.com. DOI 10.1002/hc.21034
Co-reporter:Lu-Jie Cao, Hong-Qi Ai, Li-Ming Zheng, Su-Na Wang, Mei-Juan Zhou, Ji-Feng Liu, Chong Zhang
Journal of Molecular Structure: THEOCHEM 2010 Volume 948(1–3) pp:65-70
Publication Date(Web):30 May 2010
DOI:10.1016/j.theochem.2010.02.021
The interactions of neutral and charged (−2, −1, +1, and +2) Tin clusters (n = 1–7) with one N2 molecule were investigated by density functional theory. For neutral TinN2 clusters, the nitrogen molecule is geometrically dissociative and each separated N atoms is favored to bind at the threefold hollow site for n ⩾ 3. The adsorption energies of neutral TinN2 clusters which increase monotonously from n = 1 to 4 and then oscillate from n = 4 to 7, are all energetically favorable, suggesting the higher stability of large-sized neutral TinN2 clusters. The detachment of Tix (1 ⩽ x ⩽ n − 1, n = 2–7) sub-unit from neutral TinN2 (n = 2–7) clusters are energetically unfavorable, further suggesting their thermodynamic stability. The addition of different charges (−2, −1, +1, +2) on the most stable neutral TinN2 (n = 1–7) clusters would induce their geometrical perturbations. The stabilities of neutral TinN2 systems are enhanced by adding −1 e, while would be reduced by −2, +1, and +2 e. The calculated adsorption energies of charged TinN2 clusters have the following order: TinN22+>TinN21+>TinN22->TinN21-.
Co-reporter:S.K. Xing, C. Zhang, H.Q. Ai, Q. Zhao, Q. Zhang, D.Z. Sun
Journal of Molecular Liquids 2009 Volume 146(1–2) pp:15-22
Publication Date(Web):31 May 2009
DOI:10.1016/j.molliq.2009.01.005
The inclusion process of 2′-hydroxyl-5′-methoxyacetophone (Hma) with β-cyclodextrin (β-CD), as well as their other seven possible interaction types, was investigated theoretically. The data suggest that: (1) the inclusion complex formed by Hma entering into the cavity of β-CD from its wide side (the secondary hydroxyl group side) is more stable than that from its narrow side (the primary hydroxyl group side); (2) the formation of the inclusion complex is predicted to be an enthalpy-driven process in gas phase and an enthalpy–entropy co-driven process in aqueous solution, which is in accord with the experimental results; (3) other different interaction types between Hma and β-CD should be also possibly found experimentally due to their negative binding energy (ΔE) though their distributions differ greatly. At last, comparative study of the interactions of β-CD with Hma and its two isomers, paeonol (Pae) and acetovanillone (Ace), are investigated and their obvious differences in binding energy and enthalpy change suggest that the β-CD could identify the three isomers.