Co-reporter:B. Xu;B. C. Pan
The Journal of Physical Chemistry C January 15, 2009 Volume 113(Issue 2) pp:567-570
Publication Date(Web):2017-2-22
DOI:10.1021/jp807477a
The interaction between Ga fragments and the inner wall of carbon nanotube (CNT) is studied by using the density functional theory calculations. We find that the encapsulated Gan (n = 3, 4, 5) fragments prefer to form the planar configurations. Such a planar fragment interacts with the perfect inner wall very weakly, but bonds with the monovacancy of the CNT significantly. In the latter case, there is only one Ga atom in a fragment bonding with the monovacancy, and the formed Ga−C bonds is stronger than the Ga−Ga bonds of the fragment. As a consequence, the Ga fragment may keep sliding in the inner wall of a CNT through leaving a Ga atom at the monovacancy. According to this, we suggest that although the existence of the monovacancy in CNT affects the motion of the encapsulated Ga nanowire, the Ga nanowire encapsulated can slides in the inner wall of the defected CNT easily when the monovacancies in the CNT are dilute.
Co-reporter:Fanghua Ning;Jing Shi;Haibin Su;Musheng Wu;Gang Liu;Chuying Ouyang
Journal of Materials Chemistry A 2017 vol. 5(Issue 20) pp:9618-9626
Publication Date(Web):2017/05/23
DOI:10.1039/C7TA01339F
The migration of Li ions in electrode materials is the main limiting factor to determine the rate capability of Li-ion batteries. In this work, the influences of Jahn–Teller (JT) distortion on Li migration in full delithiated LiMn2O4 (λ-MnO2) were systematically studied by a first-principles computational approach. Our results unravel the direction of JT distortion strongly affecting the activation barrier of Li-ion migration in λ-MnO2. In particular, Li-ion migration has the lowest activation barrier when the two elongated Mn–O bonds of Mn3+ ion are quasi-collinear with the linked Li–O bonds in the transition state, in contrast with the highest barrier when the elongated Mn–O bonds are approximately perpendicular to the linked Li–O bonds. In addition, lattice-strain-induced variation of the Li-migration barrier in λ-MnO2 exhibits either upward or downward trends, depending on the detailed coupling with JT distortion. Further analysis showed that the difference in activation barriers can be explained by the different Li–O distances in terms of the Coulomb interaction energies, which is induced by the different position and direction of JT distortion. Finally, the Li-ion migration in the whole λ-MnO2 system is also discussed by considering the influences of JT polarons.
Co-reporter:Fanghua Ning, Bo Xu, Jing Shi, Musheng Wu, Yinquan Hu, and Chuying Ouyang
The Journal of Physical Chemistry C 2016 Volume 120(Issue 33) pp:18428-18434
Publication Date(Web):August 2, 2016
DOI:10.1021/acs.jpcc.6b05091
Rare earth elements, known for their large radius, high charge, and strong self-polarization ability, are expected to bring improvements in Li-ion batteries. However, some basic issues such as structural variation, the nature of the improved Li mobility, and electronic conductivity in rare earth-doped electrode compounds are still unrevealed. In the present work, the structural, electronic, and Li migration properties of Ce- and La-doped LiCoO2 cathode materials are systematically studied by using the first-principles calculations. The results show that after rare earth elements are doped, the cell volume expands with local structure distortion around the substitution site. Meanwhile, the doped systems remain insulating characteristics with decreased band gap. The migration barriers vary considerably depending on different paths due to the competition between the increase of Li slab distance and the variation of potential energy surface caused by the doping of rare earth elements. The minimum activation barriers for Li motion decease significantly from 0.669 to 0.382 eV and to 0.239 eV upon Ce and La doping, respectively. Furthermore, Li ion migrations along the entire supercell are also studied.
Co-reporter:Taoyuan Du
The Journal of Physical Chemistry C 2016 Volume 120(Issue 11) pp:5876-5882
Publication Date(Web):March 1, 2016
DOI:10.1021/acs.jpcc.5b11891
Co-reporter:Junping Hu, Bo Xu, Shengyuan A. Yang, Shan Guan, Chuying Ouyang, and Yugui Yao
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 43) pp:24016
Publication Date(Web):October 13, 2015
DOI:10.1021/acsami.5b06847
Searching for suitable anodes with good performance is a key challenge for rechargeable Na-ion batteries (NIBs). Using the first-principles method, we predict that 2D nitrogen electride materials can be served as anode materials for NIBs. Particularly, we show that Ca2N meets almost all the requirements of a good NIB anode. Each formula unit of a monolayer Ca2N sheet can absorb up to four Na atoms, corresponding to a theoretical specific capacity of 1138 mAh·g–1. The metallic character for both pristine Ca2N and its Na intercalated state NaxCa2N ensures good electronic conduction. Na diffusion along the 2D monolayer plane can be very fast even at room temperature, with a Na migration energy barrier as small as 0.084 eV. These properties are key to the excellent rate performance of an anode material. The average open-circuit voltage is calculated to be 0.18 V vs Na/Na+ for the chemical stoichiometry of Na2Ca2N and 0.09 V for Na4Ca2N. The relatively low average open-circuit voltage is beneficial to the overall voltage of the cell. In addition, the 2D monolayers have very small lattice change upon Na intercalation, which ensures a good cycling stability. All these results demonstrate that the Ca2N monolayer could be an excellent anode material for NIBs.Keywords: 2D electride; anode material; diffusion barrier; Na-ion batteries; open-circuit voltage
Co-reporter:B. Xu, L. Wang, H.J. Chen, J. Zhao, G. Liu, M.S. Wu
Computational Materials Science 2014 Volume 93() pp:86-90
Publication Date(Web):October 2014
DOI:10.1016/j.commatsci.2014.06.033
•The adsorption and diffusion of Li on the 1T-MoS2, a metastable phase, is studied by using DFT calculations.•It is found that the binding energy decreases with the increase of the Li concentration.•The electronic structure of Li-adsorbed MoS2 is studied.•The diffusion energy barrier of Li atom is related with the Li concentration.•1T-MoS2 could be used as electrode material in Li ion batteries.Using first-principles calculations, we investigate the Li adsorption and diffusion on the 1T-MoS2 monolayer. Our calculations demonstrate that the binding energy decreases with the increase of the Li concentration, regardless of adsorption on one side or two sides. The binding energy corresponding to the case of Li:Mo = 2:1 is still higher than the cohesive energy of metal Li. The diffusion energy barrier of Li atom is closely related with the Li concentration, larger than that of Li on 1H-MoS2 but comparable to that of LiFePO4. Our calculations suggest that 1T-MoS2 monolayer could be a promising electrode material for lithium ion batteries in terms of lithium storage capacity and diffusion kinetics.Graphical abstractHigh binding energy and medium diffusion barrier for lithium on 1T-MoS2 monolayer.
Co-reporter:B. Xu, M.S. Wu, G. Liu, X.L. Lei, J. Ouyang, C.Y. Ouyang
Physica E: Low-dimensional Systems and Nanostructures 2014 Volume 58() pp:153-156
Publication Date(Web):April 2014
DOI:10.1016/j.physe.2013.12.013
Highlights•The electronic and magnetic properties of the zigzag (n , 0) (n=6–10)(n=6–10) SiC/C nanotube superlattices are investigated by using the first-principles calculations.•The (6, 0) SiC/C nanotube superlattice shows magnetic property with 2.0μB2.0μB magnetic moments due to the curvature effect.•The spin densities distribute periodically along the axial direction and mainly locate at the CNT segment, thus forming the magnetic nanonodes.The zigzag (n, 0) (n=6–10) SiC/C nanotube superlattices are investigated by using the first-principles calculations. Our calculations indicate that the (n, 0) (n =7–10) SiC/C nanotube superlattices are semiconducting and non-magnetic. However, the (6, 0) SiC/C nanotube superlattice shows magnetic property with 2.0μB2.0μB magnetic moments due to the curvature effect. The spin densities distribute periodically along the axial direction and mainly locate at the CNT segment, thus forming the magnetic nanonodes. The proposed magnetic nanonodes offer a method to obtain the periodic magnetic structure and thus have potential applications in future nanoscale devices.
Co-reporter:Junping Hu ; Bo Xu ; Chuying Ouyang ; Shengyuan A. Yang ;Yugui Yao
The Journal of Physical Chemistry C 2014 Volume 118(Issue 42) pp:24274-24281
Publication Date(Web):September 25, 2014
DOI:10.1021/jp507336x
First-principles calculations are performed to study the electronic properties and Li storage capability of V2C and its corresponding fluoride and hydroxide. We find that the V2C monolayer is metallic with antiferromagnetic configuration, while its derived V2CF2 and V2C(OH)2 in their the most stable configurations are small-gap antiferromagnetic semiconductors. Li adsorption could enhance the electric conductivity of V2C fluoride and hydroxide. The bare V2C monolayer shows fast Li diffusion with low diffusion barrier height and very high Li storage capacity (with theoretical value ∼940 mAh/g), while the passivated F or OH atoms on its surface tend to impede Li diffusion and largely reduce the Li storage capacity. Moreover, the average intercalation potentials for V2C-based materials are calculated to be relatively low. Our results suggest that V2C monolayer could be a promising anode material for Li-ion batteries.
Co-reporter:Fanghua Ning, Bo Xu, Jing Shi, Haibin Su, Musheng Wu, Gang Liu and Chuying Ouyang
Journal of Materials Chemistry A 2017 - vol. 5(Issue 20) pp:NaN9626-9626
Publication Date(Web):2017/04/14
DOI:10.1039/C7TA01339F
The migration of Li ions in electrode materials is the main limiting factor to determine the rate capability of Li-ion batteries. In this work, the influences of Jahn–Teller (JT) distortion on Li migration in full delithiated LiMn2O4 (λ-MnO2) were systematically studied by a first-principles computational approach. Our results unravel the direction of JT distortion strongly affecting the activation barrier of Li-ion migration in λ-MnO2. In particular, Li-ion migration has the lowest activation barrier when the two elongated Mn–O bonds of Mn3+ ion are quasi-collinear with the linked Li–O bonds in the transition state, in contrast with the highest barrier when the elongated Mn–O bonds are approximately perpendicular to the linked Li–O bonds. In addition, lattice-strain-induced variation of the Li-migration barrier in λ-MnO2 exhibits either upward or downward trends, depending on the detailed coupling with JT distortion. Further analysis showed that the difference in activation barriers can be explained by the different Li–O distances in terms of the Coulomb interaction energies, which is induced by the different position and direction of JT distortion. Finally, the Li-ion migration in the whole λ-MnO2 system is also discussed by considering the influences of JT polarons.












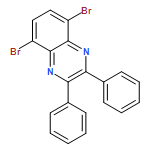
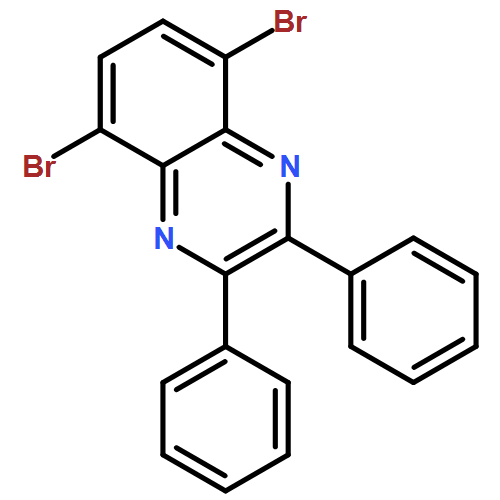
![Pyrido[3,4-b]pyrazine, 5,8-dibromo-2,3-diphenyl-](/data/chemimg/3110200/221241-48-1.png)
![Pyrido[3,4-b]pyrazine, 5,8-dibromo-2,3-diphenyl-](/data/chemimg/3110200/221241-48-1_b.png)




![2,3-diphenyl-Pyrido[3,4-b]pyrazine](http://img.cochemist.com/ccimg/67900/67899-59-6.png)
![2,3-diphenyl-Pyrido[3,4-b]pyrazine](http://img.cochemist.com/ccimg/67900/67899-59-6_b.png)