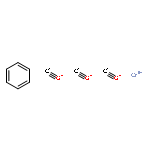
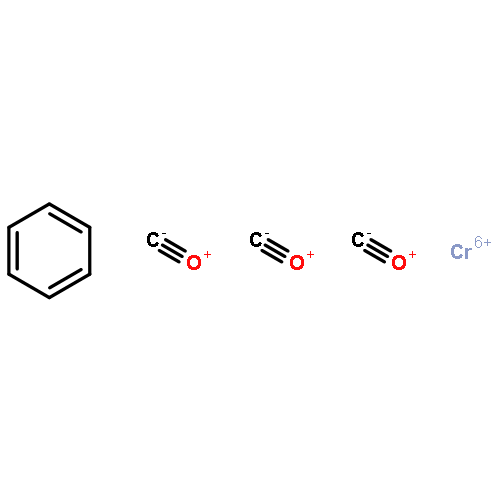
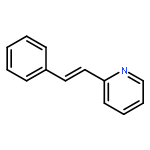
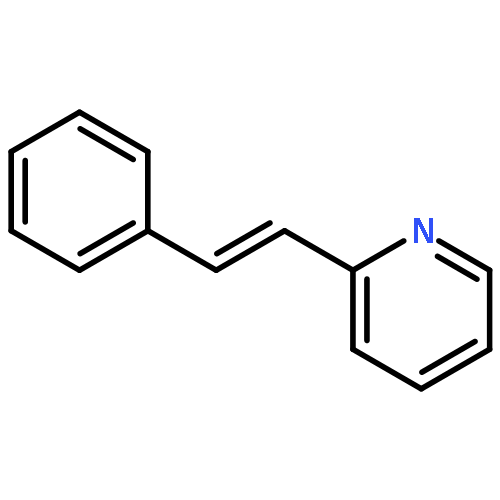
Co-reporter:Kristy M. DeWitt, Tung T. To, Edwin J. Heilweil, and Theodore J. Burkey
The Journal of Physical Chemistry B 2015 Volume 119(Issue 17) pp:5531-5536
Publication Date(Web):March 25, 2015
DOI:10.1021/jp513033j
The extent of the photoinitiated linkage isomerization of dicarbonyl(3-cyanomethylpyridine-κN)(η5-methylcyclopentadienyl)manganese (4) to dicarbonyl(3-cyano-κN-methylpyridine)(η5-methylcyclopentadienyl)manganese (5) was examined by time-resolved infrared spectroscopy on picosecond to microsecond time scales in room temperature isooctane to determine the extent the isomerization occurs as a geminate cage rearrangement. We previously reported that a substantial part of the conversion between 4 and 5 must be a bimolecular reaction between a solvent coordinated dicarbonyl(η5-methylcyclopentadienyl)manganese (3) and uncoordinated 3-cyanomethylpyridine. For the purpose of designing a molecular device, it would be desirable for the photoisomerization to occur in a geminate cage reaction, because the faster the isomerization, the less opportunity for side reactions to occur. In this study, assignments of transients are identified by comparison with transients observed for model reactions. Within 100 μs after photolysis of 4 in isooctane, no 5 is observed. Instead, the solvent coordinated 3 is observed within 25 ps after irradiation. The formation of 5 is observed only in the presence of 9 mM 3-cyanomethylpyridine but not until 10–50 μs after irradiation of 4. Within the limits of detection, these results indicate the conversion of 4 to 5 occurs exclusively via a bimolecular reaction of 3-cyanomethylpyridine with solvent coordinated 3 and not a geminate cage reaction between 3-cyanomethylpyridine and the dicarbonyl(η5-methylcyclopentadienyl)manganese fragment.
Co-reporter:Roger G. Letterman, Charles B. Duke III, Tung T. To, Theodore J. Burkey, and Charles Edwin Webster
Organometallics 2014 Volume 33(Issue 21) pp:5928-5931
Publication Date(Web):October 8, 2014
DOI:10.1021/om5007165
Competing degenerate pathways for ring inversion in organometallic complexes are proposed to be ubiquitous examples that adhere to the principle of microscopic reversibility. The NMR spectra for ring inversion of two chromium arene dicarbonyl pyridyl chelates ([Cr{η6-C6H5(CH2)n(2-Py-κN)}(CO)2]; 2-Py = 2-pyridyl, n = 2 (1), and 3 (2)) and a manganese cyclopentadienyl dicarbonyl methyl sulfide chelate ([Mn{η5-C5H4COC(SCH3)2(SCH3-κS)}(CO)2] (3)) were characterized via variable-temperature NMR spectroscopy and DFT theoretical calculations.
Co-reporter:Charles B. Duke III, Roger G. Letterman, Jermaine O. Johnson, James W. Barr, Songnan Hu, Charles R. Ross II, Charles Edwin Webster, and Theodore J. Burkey
Organometallics 2014 Volume 33(Issue 2) pp:485-497
Publication Date(Web):January 14, 2014
DOI:10.1021/om400928k
Chromium arene tricarbonyl complexes with tethered pyridinyl groups [Cr{η6-C6H5(CH2)n(2-Py)}(CO)3] (4–6) (2-Py = 2-pyridinyl, n = 1–3, respectively) were synthesized and irradiated to form the chelates [Cr{η6-C6H5(CH2)n(2-Py)-κN}(CO)2] (7–9). Studies examined the effect of ring size and structure on chromophore λmax, stability, and photosensitivity, which are factors important for photochromes based on linkage isomerization of tethered functional groups. The studies also include [Cr{η6-C6H5CH(2-Py)CH2CH═CH2}(CO)3] (3), which has a bifunctional tether of propenyl and pyridinyl groups, and irradiation produces the linkage isomers [Cr{η6-C6H5(CH(2-Py)CH2CH═CH2)-κN}(CO)2] (1) and [Cr{η6-C6H5(CH(2-Py)CH2CH═CH2)(η2-CH═CH2)}(CO)2] (2). X-ray crystal structures for 7–9 show that the dihedral angle between the coordinated pyridinyl groups and the phenyl-chromium centroid increases from 1 to 73° (n = 1–3, respectively). The experimental and TDDFT computed optical changes accompanying an increase in the dihedral angle are modest and not monotonic for 7–9 due to structural changes inherent in the chelate rings. An increase in Cr–N bond lengths and decrease in their bond energies were observed experimentally and computationally for the series of 7–9. The quantum yields for formation of the five-, six-, and seven-membered chelate rings during the conversion of 4–6 to 7–9, respectively, were within experimental error for that observed for conversion of 10 [Cr{η6-C6H6}(CO)3] with free pyridine to 11 [Cr{η6-C6H6}(C5H5N-κN)(CO)2], indicating that the product-determining step precedes chelation. The enthalpies for chelation of 4–6 to 7–9 were determined independently by photoacoustic calorimetry and DFT computations. The computationally derived mechanism for thermal isomerization of 1 to 2 indicates that the transition state is a dissociative interchange with a free energy of activation of 27.9 kcal mol–1 (1 → 2), a result consistent with an experimentally bistable photochrome. The results indicate which tether properties are important for optimizing photochrome performance.
Co-reporter:Shannon M. Gittermann, Roger G. Letterman, Tianjie Jiao, Ging-Long Leu, Nathan J. DeYonker, Charles Edwin Webster, and Theodore J. Burkey
The Journal of Physical Chemistry A 2011 Volume 115(Issue 32) pp:9004-9013
Publication Date(Web):July 22, 2011
DOI:10.1021/jp203915q
The photosubstitution reactions of molybdenum hexacarbonyl with σ and π donor ligands were investigated using photoacoustic calorimetry and computational methods in a series of linear alkane solvents (pentane, hexane, heptane, octane, decane, and dodecane). The results show that reaction volumes make a significant contribution to the photoacoustic signal and must be considered during thermodynamic calculations based on photoacoustic measurements. The enthalpies of CO substitution by an alkane solvent and subsequent substitution by each Lewis base were determined. Corresponding Mo–L bond energies (kcal mol–1) were calculated: L = linear alkanes (13), triethylsilane (26), 1-hexyne (27), 1-hexene (27), and benzene (17). The relative energies are in agreement with computational results. The experimental reaction volume for CO substitution by alkane was positive (15 mL mol–1) and negative or close to zero for alkane substitution by a Lewis base (for example, −11 mL mol–1 for triethylsilane and 3.6 mL mol–1 for benzene). The errors in the experimental and computational reaction volumes are large and often comparable to the reaction volumes. An improved calibration of the methods as well as a better understanding of the underlying physics involved is needed. For the Lewis bases reported in this study, the second-order rate constants for the displacement of a coordinated alkane are less than diffusion control (5 × 106–4 × 107 M–1 s–1) and decrease monotonically with the alkane chain length. The rate constants correlate better with steric effects than with bond energies. An interchange mechanism is consistent with the results.
Co-reporter:Edwin J. Heilweil, Jermaine O. Johnson, Karen L. Mosley, Philippe P. Lubet, Charles Edwin Webster, and Theodore J. Burkey
Organometallics 2011 Volume 30(Issue 21) pp:5611-5619
Publication Date(Web):October 24, 2011
DOI:10.1021/om2003656
The chelation following photodissociation of CO for cyclopentadienyl manganese tricarbonyl derivatives with a bifunctional side chain has been investigated. Previous studies show that steady-state irradiation of 1 (Mn{η5-C5H4CH2COR}(CO)3, R = 2-pyridyl) leads to CO dissociation and formation of O-chelate 2 with smaller amounts of N-chelate 3. Subsequently, 2 rearranges thermally to 3. A new preparation for 1 is reported, while analogues 4 (R = phenyl) and 5 (R = 4-pyridyl) are prepared for the first time. Steady-state UV–vis, FTIR, and NMR studies of 4 and 5 in heptane demonstrate that O-chelates 6 and 7, respectively, are formed with the side-chain oxygen but decay on the minute time scale. The linkage isomerization of O-chelate 2 to 3 is faster than the decay observed for the O-chelate 6 (R = 2-pyridyl versus Ph), even in the presence of 0.1 M pyridine for the latter. Following irradiation of 4 during time-resolved infrared studies in heptane, ultrafast O-chelation is observed but not ultrafast solvent coordination. Ultrafast O-chelation is also observed for 5 along with an unidentified transient. Following irradiation of 1, ultrafast O- and N-chelation are observed, to the exclusion of ultrafast solvent coordination. This result suggests that chelate formation is a subpicosecond process and that both chelates are formed independently. A split in the otherwise degenerate stretching bands for 4 and 5 in FTIR spectra suggests that there is significant electronic communication between the side chain and the metal carbonyl groups. The results suggest that ultrafast chelation is favored by side-chain conformations that position a functional group near the metal center.
Co-reporter:Tung T. To and Edwin J. Heilweil, Charles B. Duke III, Kristie R. Ruddick, Charles Edwin Webster and Theodore J. Burkey
The Journal of Physical Chemistry A 2009 Volume 113(Issue 12) pp:2666-2676
Publication Date(Web):February 20, 2009
DOI:10.1021/jp8068909
We review recent studies of processes relevant to photoinduced linkage isomerization of organometallic systems with the goal of preparing organometallics with an efficient and ultrafast photochromic response. The organometallic system thus corresponds to two linkage isomers with different electronic environments that are responsible for different optical properties. Much of this work has focused on examining processes following irradiation of cyclopentadienyl manganese tricarbonyl derivatives (compounds 3−21) including solvent coordination, thermal relaxation, solvent displacement by tethered functional groups (chelation), dissociation of tethered functional groups, and linkage isomerization. A new platform is investigated for obtaining a photochromic response in new experiments with arene chromium dicarbonyl complexes. A photochromic response is observed for arene chromium dicarbonyl complexes with tethered pyridine and olefin functional groups based on light-driven linkage isomerization on the nanosecond time scale. Irradiation at 532 nm of 23 ([Cr{η6-C6H5CH(2-Py-κN)CH2CH═CH2}(CO)2]) (Py = pyridine) results in the isomerization to 22 ([Cr{η6-C6H5CH(2-Py)CH2-η2-CH═CH2}(CO)2]), and 355 nm irradiation isomerizes 22 to 23. The ultrafast linkage isomerization has been investigated at room temperature in n-heptane solution on the picosecond to microsecond time scale with UV- or visible-pump and IR-probe transient absorption spectroscopy by comparing the dynamics with model compounds containing only a tethered pyridine. Irradiation of 24 ([Cr{η6-C6H5(CH2)3(2-Py)}(CO)3]) and 25 ([Cr{η6-C6H5(CH2)2(2-Py)}(CO)3]) at 289 nm induces CO loss to immediately yield a Cr−heptane solvent coordinated intermediate of the unsaturated Cr fragment, which then converts to the κN1-pyridine chelate within 200 and 100 ns, respectively. Irradiation of 26 ([Cr{η6-C6H5CH2(2-Py)}(CO)3]) also induces CO loss to immediately yield three species: the Cr−heptane solvent coordinated intermediate, a κN1-Py nitrogen chelate, and an agostic η2-chelate in which the pyridine is coordinated to the metal center via a C−H agostic bond as opposed to the nitrogen lone pair. Both the transient Cr−heptane coordinated intermediate and the agostic pyridine chelate convert to the stable κN1-pyridine chelate within 50 ns. Similar reaction dynamics and transient species are observed for the chelate 33 ([Cr{η6-C6H5CH2(2-Py)-κN}(CO)2]) where a Cr−Py bond, not a Cr−CO bond, initially cleaves.
Co-reporter:Tung T. To, Charles B. Duke III, Christopher S. Junker, Casey M. O’Brien, Charles R. Ross II, Craig E. Barnes, Charles Edwin Webster and Theodore J. Burkey
Organometallics 2008 Volume 27(Issue 2) pp:289-296
Publication Date(Web):January 3, 2008
DOI:10.1021/om701101h
Two, bifunctional side-chain cyclopentadienylmanganese tricarbonyl complexes, 7 (pyridine and ketone side chain) and 14 (thioamide and pyridine side chain), were prepared and converted to chelates following CO dissociation by UV irradiation. Both pyridine (8) and carbonyl (9) chelates are observed following irradiation of 7. In contrast, only thioamide chelate (16) is observed following irradiation of 14 even though a pyridine group was available. Visible irradiation isomerizes the pyridine chelate 8 to the carbonyl chelate 9, which thermally isomerizes back to 8 at 25 °C in a few minutes, demonstrating a photochromic response from a cyclopentadienyl-manganese complex based on a linkage isomerization of a tethered functional group. DFT calculations predicted that the activation enthalpy of thermal linkage isomerization would be 20.8 kcal/mol and that the mechanism is likely an associative process that does not involve a simple dissociation of the manganese bond to the side-chain ketone. The DFT calculations were supported by subsequent dynamic NMR experiments that yielded an activation enthalpy and entropy of 21.4 ± 0.8 kcal/mol and 3.5 ± 0.1 eu, respectively. The studies indicate that a compound with a tethered, coordinated functional group, which is otherwise not labile, can isomerize by a low-energy pathway if there is an appropriate “conduit” to another functional group with a stronger metal–ligand bond. Thus, the preparation of photochromic organometallic complexes based on linkage isomerization will require a bridge that inhibits an associative walk between functional groups if they are to be bistable.
Co-reporter:Gerard J. Farrell, Theodore J. Burkey
Journal of Photochemistry and Photobiology A: Chemistry 2000 Volume 137(2–3) pp:135-139
Publication Date(Web):4 December 2000
DOI:10.1016/S1010-6030(00)00371-3
We have developed high-pressure photoacoustic calorimetry to determine the volume of reaction for organometallic compounds. This allows the calculation of reaction enthalpies from the photoacoustic data. The photoacoustic signal for chromium hexacarbonyl with acetonitrile in heptane was examined from near ambient pressure to 100 MPa. Two components of the signal were resolved at 10–20 mM acetonitrile. The fast component is attributed to CO displacement by heptane (<10 ns), while the slow component (∼1 μs) is attributed to the subsequent displacement of heptane by acetonitrile. The amplitude of both components were pressure dependent demonstrating that the volumes of each reaction made a significant contribution to the photoacoustic signal (−21 and 17 ml mol−1, respectively). The enthalpies of CO displacement by heptane are 28 kcal mol−1 and heptane displacement by acetonitrile is −22 kcal mol−1. Neglect of the volumes of reaction in the PAC experiments can lead to an error of 6 kcal mol−1.