Co-reporter:Congting Sun, Xiaoyan Chen, and Dongfeng Xue
Crystal Growth & Design May 3, 2017 Volume 17(Issue 5) pp:2631-2631
Publication Date(Web):April 13, 2017
DOI:10.1021/acs.cgd.7b00145
Structurally, hydrogen bonding is identified as a key factor to domain the construction of a crystallographic frame during the crystallization of the Ln(H2O)9(CF3SO3)3 (Ln = La–Lu) system. In situ Raman spectroscopy is used to capture the hydrogen bonding dependent mesoscale frameworks that are formed during Ln(H2O)9(CF3SO3)3 crystallization in aqueous solution by continuously collecting the spectra of structural fragments. The spectral characteristics show that the isolated Ln(H2O)93+ tricapped trigonal prisms cannot exist in the aqueous solution. With the concentration of aqueous solution, the hydrated Ln3+ and CF3SO3– tend to share common H2O molecules, and new hydrogen bonding will be built surrounding Ln3+. Especially, for the Nd, Eu, Yb, and Lu system, Ln(H2O)n(CF3SO3)3 (n = 8–9) clusters instead of hydrated Ln3+ and CF3SO3– are formed in the solution. Under the guiding of intermolecular hydrogen bonds, both bond lengths and bond angles of Ln–O may be regulated, leading to the initial formation of Ln(H2O)63+ prisms and the following Ln(H2O)93+ tricapped trigonal prisms. Meanwhile, the symmetry of both CF3 and SO3 groups decreases from C3h to C2 accompanied by the formation of Ln(H2O)63+ triprism. The present study opens up the chemical bonding behaviors of rare earth ions in aqueous solution, which provides basic data for the study of the coordination of rare earth complexes and the design of novel rare earth materials.
Co-reporter:Kunfeng Chen, Dongfeng Xue, Sridhar Komarneni
Journal of Colloid and Interface Science 2017 Volume 487() pp:156-161
Publication Date(Web):1 February 2017
DOI:10.1016/j.jcis.2016.10.028
Nanoclay assisted electrochemical exfoliation was developed to in-situ form functionalized graphene electrode materials from pencil core with different ratios of graphite and clay. This method made a positive transformation from solid graphite to graphene colloidal solution, which can be used to construct binder- and additive-free thin-film electrodes. Exfoliated graphene can be served as both conductive current collector (film resistance of 33 Ω/square) and electrode materials. Graphene thin-film electrodes from pencil cores displayed higher capacity of 224 than 80 mA h/g of that from pure graphite. The electrochemical performance can be controlled by the ratio of graphite and clay and the oxidation reaction of surface oxygen functional groups. The described nanoclay-assisted electrochemical oxidation route shows great potential for the synthesis of functionalized graphene electrode materials for high-conductive thin-film lithium ion batteries and supercapacitors.High conductive graphene colloidal inks were synthesized by nanoclay assisted electrochemical exfoliation of pencil core, which can be easily used to construct high-conductive thin-film electrodes.
Co-reporter:Dongfeng Xue;Congting Sun;Xiaoyan Chen
Chinese Journal of Chemistry 2017 Volume 35(Issue 9) pp:1452-1458
Publication Date(Web):2017/09/01
DOI:10.1002/cjoc.201700425
Both the bonding mode and geometry can serve as the chemical bonding nature of central cation, which is essentially determined by the atomic orbital-hybridization. In this work, we focus on the possible chemical bonding scheme of central cations on the basis of a quantitative analysis of electron domain of an atom. Starting from the hybridization of outer atomic orbitals that are occupied by valence electrons, we studied the possible orbital hybridization scheme of atoms in the periodic table and the corresponding coordination number as well as possible molecular geometries. According to distinct hybrid orbital sets, the chemical bonding of central cations can be classified into three typical types, resulting in the cations with a variety of coordination numbers ranging from 2 to 16. Owing to different hybridization modes, the highest coordination number of cations in IA and IIA groups is larger than that in IB-VIIIB groups, and the coordination number of lanthanide elements is most abundant. We also selected NaNO3, Fe(NO3)3•9H2O, Zn(NO3)2•6H2O, Y(NO3)3•3H2O, and La(NO3)3•6H2O as examples to confirm the direct relationship between chemical bonding characteristics and orbital hybrid set by IR spectra. The present study opens the door to reveal the chemical bonding nature of atoms on the basis of hybridization and will provide theoretical guides in structural design at an atomic level.
Co-reporter:DongFeng Xue;CongTing Sun
Science China Technological Sciences 2017 Volume 60( Issue 11) pp:1767-1768
Publication Date(Web):17 August 2017
DOI:10.1007/s11431-017-9112-1
Co-reporter:Dongfeng Xue, Hongjie Zhang, Yinong Liu, Shu Yin, Li Lu, Jin-Hyo Boo
Materials Research Bulletin 2017 Volume 96, Part 1(Volume 96, Part 1) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.materresbull.2017.09.047
Co-reporter:Kunfeng Chen
Chinese Journal of Chemistry 2017 Volume 35(Issue 6) pp:861-866
Publication Date(Web):2017/06/01
DOI:10.1002/cjoc.201600785
AbstractBiomass-derived activated carbon electrode materials have been synthesized by carbonization and KOH activation processes from an agriculture waste − rice husk, composed of organic compound and silica. The surface area of activated carbon reached 1098.1 m2/g mainly including mesopores and macropores due to the template effect of silica in rice husk. Owing to the existence of mesopores and macropores, the as-obtained activated carbon materials can be used in aqueous supercapacitors, lithium-ion (Li-ion) capacitors and lithium-sulfur (Li-S) batteries. In KOH electrolyte, fast rate performance (as high as 2 V/s) was obtained due to the existence of ideal electrical double layer capacitance. In organic electrolyte, high voltage (2.5 V) was achieved. Activated carbon electrode for Li-ion capacitor also showed capacity of 17 mAh/g at 100 mA/g with the high voltage range of 2.5 V. The capacities of sulfur-activated carbon in Li-S batteries were 1230 and 970 mAh/g at the current densities of 0.1 and 0.2 C. The present results showed that activated carbon materials with mesopores were good host to immobilize polysulfides.
Co-reporter:Congting Sun, Dongfeng Xue
Journal of Crystal Growth 2017 Volume 470(Volume 470) pp:
Publication Date(Web):15 July 2017
DOI:10.1016/j.jcrysgro.2017.04.009
•Phase transition of materials components drives crystallization process.•Chemical potential decrease is identified as a key parameter in crystallization.•Chemical potential decrease in crystal growth is scaled by chemical bonding model.•Different chemical bonding architectures at interface produce distinct geometries.A chemical bonding model is established to describe the chemical potential decrease during crystallization. In the nucleation stage, in situ molecular vibration spectroscopy shows the increased vibration energy of constituent groups, indicating the shortened chemical bonding and the decreased chemical potential towards the formation of nuclei. Starting from the Gibbs free energy formula, the chemical potential decrease during crystallization is scaled, which depends on the released chemical bonding energy per unit phase transition zone. In the crystal growth, the direction-dependent growth rate of inorganic single crystals can be quantitatively determined, their anisotropic thermodynamic morphology can thus be constructed on the basis of relative growth rates.
Co-reporter:Kunfeng Chen, Dongfeng Xue
Materials Research Bulletin 2017 Volume 96, Part 3(Volume 96, Part 3) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.materresbull.2017.01.025
•Graphene was synthesized by electrochemical exfoliation of graphite-clay rods with different clay:graphite ratios.•In-situ electrochemical oxidation and functionalization were used in the exfoliated process.•Graphene thin-film electrodes were constructed for supercapacitors and lithium ion batteries.•This work provided a value-added way that can transform the low-purity graphite resources to graphene electrode materials.The development of cost- and time-effective methods to synthesize graphene materials is very urgent in order to promote its large-scale application in electrochemical energy storage devices. Herein, graphene materials were synthesized by the electrochemical exfoliation (electrochemical oxidation) of graphite-clay rods with different clay:graphite ratios in aqueous solution and in short reaction time (1 h). The dual role of electrochemical oxidation reaction was presented: (1) the electrochemical oxidation reaction facilitates the exfoliation of pencil graphite leads and in-situ forms functional groups on graphene; (2) the electrochemical oxidation reaction enhances the Faradaic reaction in supercapacitor. The in-situ electrochemical oxidation and functionalization methods can transform low purity graphite to high-value graphene electrode materials for supercapacitors and lithium ion batteries. The specific capacitances of exfoliated graphene are 20-time larger than that of graphite-clay composites. This work provided a value-added way that can transform the low-purity graphite resources to high performance graphene electrode materials.Download high-res image (161KB)Download full-size image
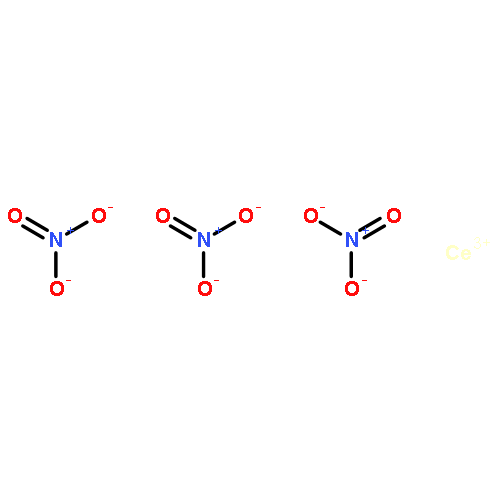
Co-reporter:Dongfeng XUE, Congting SUN, Xiaoyan CHEN
Journal of Rare Earths 2017 Volume 35, Issue 8(Volume 35, Issue 8) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/S1002-0721(17)60984-0
The chemical bonding nature of rare earth (RE) elements can be studied by a quantitative analysis of electron domain of an atom. The outer electrons of RE elements are within the valence shell 4f0–145d0–16s2, which are involved in all chemical bonding features. We in this work found that the chemical bonding characteristics of 4f electrons are a kind of hybridizations, and classified them into three types of chemical bonding of 4f0–145d0–16s2, furthermore, the coordination number ranging from 2 to 16 could thus be determined. We selected Y(NO3)3, La(NO3)3, Ce(NO3)3, YCl3, LaCl3, and CeCl3 as examples to in-situ observe their IR spectra of chemical bonding behaviors of Y3+, La3+ and Ce3+ cations, which could show different chemical bonding modes of 4f and 5d electrons. In the present study, we obtained the direct criterion to confirm whether 4f electrons can participate in chemical bonding, that is, only when the coordination number of RE cations is larger than 9.
Co-reporter:Kunfeng Chen
CrystEngComm (1999-Present) 2017 vol. 19(Issue 8) pp:1230-1238
Publication Date(Web):2017/02/20
DOI:10.1039/C6CE02462A
A general and programmed fast crystallization process (about 12 seconds) was designed to synthesize binary (MOx) and ternary (ABO3, A2BO4) metal oxide nanomaterials with controllable sizes and composition. The fast crystallization process mainly included the burning of metal-nitrate–filter-paper without additional energy supply and it can lead to the formation of exceptionally fine binary and ternary crystallites with sizes of ∼10–20 nm. The thermodynamic reduction potential of the metal-nitrate–filter-paper burning system can be estimated to be between −0.26 and 1.72 V, which can favor the occurrence of redox reactions, i.e. Cu2+ → Cu+ → Cu, Ni2+ → Ni, Co2+ → Co3+, Ce3+ → Ce4+, and Pr3+ → Pr4+. When used as anode materials for lithium-ion batteries, most of the as-burned metal oxides displayed high cycling stability. The discharge capacity of CoO nanoparticles can reach as high as 501.1 mA h g−1 after continuous 50 discharge–charge cycles at a current density of 100 mA g−1. The proposed fast crystallization route provided a versatile, facile and fast method for the synthesis of functional metal oxide nanomaterials with controllable sizes and composition.
Co-reporter:Congting Sun
Dalton Transactions 2017 vol. 46(Issue 24) pp:7888-7896
Publication Date(Web):2017/06/20
DOI:10.1039/C7DT01375B
REPO4 (RE = La, Gd, Lu, Y) serves as an excellent host lattice due to its stable physicochemical properties and optical inertia. Doping Gd3+(La3+) into LuPO4 can form mixed crystals, increasing the Gd3+(La3+) concentration will induce the phase transition from tetragonal to hexagonal lattices, and the variation of the local structure around the Ce3+ activator will influence its 5d-level position and consequently 5d → 4f radiation transition. This can be attributed to the synergy effect of rare earth ions in REPO4, however, the essential mechanism of such a synergy effect on the local structure and optical property is still poorly understood. Here, we study the synergy effect of rare earth ions on the phase transition and PL emission in a Ce3+:REPO4 system on the basis of the relationship between the composition-dependent local structure around Ce3+ and its PL emission properties from a molecular view. The competition between Lu3+ and Gd3+(La3+) in REPO4 not only influences the relative atomic position but also varies the symmetry of anion groups. Infrared absorption bands indicate that the activation of P−O bonding promotes phase transition and enhances PL emission intensity. The PL emission intensity of Ce3+ is higher in a REPO4 host with a lower site symmetry PO43− group (C2) than that with a higher site symmetry PO43− group (D2d). An increased disorder degree in Ce:GdxLu1−xPO4 mixed crystals leads to the shift of the 5d-level of Ce3+ towards a higher position, resulting in the blue shift of the PL emission wavelength. Moreover, the 5d → 4f emission of Ce3+ may also be modulated towards a larger wavelength via substituting the cation site with larger-radius cations under a particular crystallographic structure in REPO4. Our results highlight the importance of disordered local structures as well as activated anion groups in the enhanced PL emission of Ce3+ activators in a host lattice.
Co-reporter:Congting Sun
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 19) pp:12407-12413
Publication Date(Web):2017/05/17
DOI:10.1039/C7CP01112A
Crystal growth is a dynamic physicochemical process, which depends on the multi-parameter synergetic control and directly determines the crystal features such as geometry and size. In this study, both thermodynamic and kinetic factors that determine inorganic single crystal growth are integrated by focusing on the mass transfer process at an interface. For the specific growth system, the integrated parameter is then classified to extract the critical control factors in anisotropic growth. The driving force of mass transfer essentially depends on the anisotropic chemical bonding architectures, leading to different concentration gradients along various [uvw] directions. Exquisitely controlling the chemical bonding architecture can therefore be used to regulate the mass transfer process of a compound in a straightforward manner, encompassing the origin of anisotropic growth as well as a variety of geometries in the formation of a multicomponent crystal.
Co-reporter:Xiaoyan Chen;Congting Sun;Sixin Wu
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 13) pp:8835-8842
Publication Date(Web):2017/03/29
DOI:10.1039/C7CP00601B
Rare earth ions can be used to construct a variety of novel structures and are favorable to chemical bonding regulation and design. In this study, the chemical bonding paradigm between rare earth ions (Ln3+) and urea molecules in an aqueous solution can be tracked by the evolution of CO, NH2, and CN vibration bands during the urea nucleation stage. Rare earth ions such as La3+, Gd3+, and Lu3+ can manipulate the nucleation time of urea via regulating the nucleation-dependant N–CO⋯H–N hydrogen-bonding between urea molecules. Two types of chemical bondings between Ln3+ and urea molecules have been confirmed, which are Ln3+⋯OC–N and Ln3+⋯NH2−C. Compared with Ln3+⋯NH2−C, Ln3+ prefers to coordinate with the OC bond in urea. With a higher concentration of rare earth ions in the solution, some N–CO⋯H–N hydrogen bonds are broken as a consequence of the incorporation of Ln3+ into the lattice, resulting in the decreased symmetry of local urea molecules in the crystalline nuclei and the consequent Ln3+ concentration-dependent nucleation time of urea. Moreover, using the ionic electronegativity scale of Ln3+, the different effects of La3+, Gd3+, and Lu3+ on urea nucleation can be further distinguished. The present study provides basic data for unrevealing the chemical bonding regulation role of rare earth ions in the formation of hydrogen bonded materials, which may give insight into the design and fabrication of novel materials utilizing rare earth ions to adjust the chemical bonding process.
Co-reporter:Kunfeng Chen and Dongfeng Xue
Journal of Materials Chemistry A 2016 vol. 4(Issue 20) pp:7522-7537
Publication Date(Web):11 Apr 2016
DOI:10.1039/C6TA01527A
Materials chemistry focuses on all aspects of the production of electrode materials or the properties or applications of materials related to energy storage, which thus plays an important role in the field of energy storage. Electrochemical energy storage includes the conversion reaction between chemical energy and electric energy, with the electric energy being stored in chemical bonds of electrode materials of both battery and pseudocapacitor types. Energy density, power density and safety of these devices, i.e. lithium ion batteries and supercapacitors, are mostly dependent on the electrode materials with high electroactivity, high electron/ion conductivity, and high structural/electrochemical stability. Following the function-directed materials design rule, we can select appropriate elements, chemical bonds, crystal structures, and morphologies of those materials toward high electrochemical performances. In this review, we summarize, from both theoretical and experimental viewpoints of materials chemistry, recent advances in designing electrode materials from element and structure selections to final morphology selection. Electronegativity, atom radius, chemical bonding behavior, and oxidation state have been identified as controllable materials properties to synthesize high-performance electrode materials. This review provides general materials chemistry rules to rationally design electrode materials with improved electrochemical performance.
Co-reporter:Kunfeng Chen and Dongfeng Xue
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 43) pp:29522
Publication Date(Web):October 14, 2016
DOI:10.1021/acsami.6b10638
A novel and creative in-situ electrochemical activation method to transform vanadium ions to highly electroactive colloidal cathode in KOH solution under electric field has been designed. After undergoing electrochemical reaction, the in-situ-functionalized vanadium-based colloidal cathode can adapt their geometrical structure to the high pseudocapacitive activity. The vanadium-based colloids//activated carbon asymmetric supercapcitor displays a high energy density of 50.4 Wh/kg at a power density of 250 W/kg, which is higher than most reported vanadium-based supercapacitors. The main advantage of this system is that the materials synthesis and the device operation are performed in the same reactive environment. The obtained vanadium-based colloids can display high V3+ cation utilization ratios of about 100% for one-electron redox reactions. The present results highlight a new area of research on in-situ formation of reactive electrode materials under realistic environments, which can bring new chemistry and new structures of materials that are only present under the current in-situ reactive conditions.Keywords: electroactive colloid; high potential; hybrid supercapacitor; in-situ functionalization; VCl3
Co-reporter:Congting Sun, Xingxing Li, Hao Wang, and Dongfeng Xue
Inorganic Chemistry 2016 Volume 55(Issue 6) pp:2969-2976
Publication Date(Web):February 10, 2016
DOI:10.1021/acs.inorgchem.5b02860
The luminescence properties of Ce:LuPO4 depend on both the Ce3+ center and the host lattice. In this article, we studied the dependence of the luminescence properties of Ce:LuPO4 on both the doping concentration of Ce3+ and the size and morphology of the LuPO4 matrix at micro- and nanosize regimes. The crystalline behavior of Ce:LuPO4, including its size and shape, was investigated via precursor transformation crystallization. On the basis of this crystallization approach, Ce:LuPO4 hollow nanospheres, nanorods, and regular tetrahedrons were obtained. For micro- and nanostructured Ce:LuPO4, the surface-induced chemical bonding architecture can be effectively varied by controlling the size of the crystalline material and its geometry. Our experimental observations demonstrate that one-dimensional Ce:LuPO4 nanorods doped with 0.1 mol % Ce3+ possess the best performance among the as-prepared samples. The significant anisotropy of Ce:LuPO4 nanorods can result in a larger specific surface area and enhanced luminescence properties. Moreover, the improved luminescence property of Ce:LuPO4 nanostructures can also be optimized by increasing the preferential anisotropic chemical bonding architecture to regulate the 5d level of Ce3+. Our work also shows that the photoluminescence emission intensity of Ce:LuPO4 nanorods is increased as the surface area normal to their axial direction increases. From the standpoint of crystallization, the luminescence properties of Ce3+ in nano- and microsize matrixes can be well-optimized by controlling the crystalline behavior of the host lattice under proper synthesis conditions.
Co-reporter:Keyan Li, Fenfen Shua, Xinwen Guo, Dongfeng Xue
Electrochimica Acta 2016 Volume 188() pp:793-800
Publication Date(Web):10 January 2016
DOI:10.1016/j.electacta.2015.12.047
•Porous MnO@C composite was prepared by sintering the mixture of MnC2O4 and glucose.•MnO@C composite exhibits high specific capacity and excellent rate capability as Li-ion battery anodes.•The excellent electrochemical performance can be ascribed to porous structure, good crystallinity and proper amount of carbon coating.•This facile method can be applied to synthesis of other transition metal oxide@carbon electrode materials.MnC2O4 precursor was prepared by a precipitation method, and then porous MnO@C composites were obtained by mixing MnC2O4 precursor with different amounts of glucose and sintering at different temperatures. The influences of carbon content and crystallinity on the electrochemical performance of MnO@C were investigated. Galvanostatic charge–discharge test results showed that the MnO@C sample “1.2,700” has the largest specific capacity among all samples, which can reach high specific discharge capacity of 1691 mAh g−1 at the current density of 100 mA g−1 after 200 cycles. Even at the high current density of 1600 mA g−1, a remarkable discharge capacity of ∼630 mAh g−1 can still be delivered, demonstrating a good rate capability. The excellent electrochemical performance can be ascribed to porous structure, good crystallinity and proper amount of carbon coating. This facile method can be applied to the large-scale synthesis of high performance transition metal oxide@carbon composite electrode materials.
Co-reporter:Congting Sun and Dongfeng Xue
CrystEngComm 2016 vol. 18(Issue 8) pp:1262-1272
Publication Date(Web):12 Jan 2016
DOI:10.1039/C5CE02328A
The effects of crystallization on the formation of geochemical, biological, and synthetic materials have been motivating decades of research into crystal nucleation and growth processes. The development of crystal growth theories and models can deepen the understanding of physicochemical interactions during the crystal growth process, which facilitates the designing of crystallization approaches in material production. The chemical bonding theory of single crystal growth emphasizes the dominant role of dynamic chemical bonding mechanisms at the growing interfaces. In this paper, we highlight the chemical bonding theory of single crystal growth from the chemical reaction viewpoint, by focusing on the atomic level of the growing interface between the liquid and crystal phases. Using ZnO, CeO2, MnO2 and Y3Al5O12 as examples, we review some typical applications of the chemical bonding theory of single crystal growth in calculating crystal habits, evaluating crystal properties, and guiding practical single crystal growth. Microscopically speaking, the essence of crystal growth and design is to create ideal chemical bonding architectures at both the crystal surface and the growing interface via both thermodynamic and kinetic strategies.
Co-reporter:Kunfeng Chen, Dongfeng Xue
Materials Research Bulletin 2016 Volume 83() pp:201-206
Publication Date(Web):November 2016
DOI:10.1016/j.materresbull.2016.06.013
Colloidal supercapacitor electrode materials can show high specific capacitance and fast redox kinetics.Colloid has quasi-ionic state cations, short ion diffusion length and high specific surface area.Multiple-electron transferred Faradaic reactions occur within the whole colloid.All metal cations in colloidal electrode materials can occur redox reaction to store energy.Key issues for supercapacitor electrode materials are their low energy density and slow ion/electron diffusion/transfer kinetics. The formations of nanostructured materials and composite materials with blending/coating conductive additives have been developed to solve above problems. For more efficiently using electroactive metal cations in pseudocapacitance electrode materials, one kind of colloidal supercapacitor electrode materials was introduced to get both high energy and power densities. Colloidal supercapacitor electrode materials have overwhelming advantages, i.e. quasi-ionic state cations, short ion diffusion length and high specific surface area, which is a tradeoff between confined active cations and facile cation diffusion. Multiple-electron transferred Faradaic reactions can occur within the whole colloid, leading to high specific capacitance. The development of colloidal supercapacitor electrode materials can promote the advance of supercapacitors and make their energy densities larger than 100 Wh/kg or more, resulting in replacing batteries.
Co-reporter:Kunfeng Chen, Wei Pan, and Dongfeng Xue
The Journal of Physical Chemistry C 2016 Volume 120(Issue 36) pp:20077-20081
Publication Date(Web):August 25, 2016
DOI:10.1021/acs.jpcc.6b07708
Doping is one of the important methods to modify the physical and chemical properties of functional materials, which can be used to synthesize mixed ionic and electronic conducting metal oxides. Herein, the phase transformation of MnO2 from β- to α-phase has been proven by doping Ce3+ ions. With the increase of the amount of Ce3+ ions, the sizes of MnO2 nanorods were first decreased to 10–20 nm, then increased to 70 nm. The capacitive performance indicated that the specific capacitance of Ce-doped MnO2 electrode materials increased 10-fold compared with undoped MnO2, while the charge transfer resistance of Ce-doped MnO2 decreased. The present results show that rare earth ions can be used as a promising dopant to modify the crystallization behavior and electrochemical performance of MnO2 electrode materials.
Co-reporter:Kunfeng Chen, Shuyan Song, Fei Liu and Dongfeng Xue
Chemical Society Reviews 2015 vol. 44(Issue 17) pp:6230-6257
Publication Date(Web):08 Jun 2015
DOI:10.1039/C5CS00147A
There are many practical challenges in the use of graphene materials as active components in electrochemical energy storage devices. Graphene has a much lower capacitance than the theoretical capacitance of 550 F g−1 for supercapacitors and 744 mA h g−1 for lithium ion batteries. The macroporous nature of graphene limits its volumetric energy density and the low packing density of graphene-based electrodes prevents its use in commercial applications. Increases in the capacity, energy density and power density of electroactive graphene materials are strongly dependent on their microstructural properties, such as the number of defects, stacking, the use of composite materials, conductivity, the specific surface area and the packing density. The structural design of graphene electrode materials is achieved via six main strategies: the design of non-stacking and three-dimensional graphene; the synthesis of highly packed graphene; the production of graphene with a high specific surface area and high conductivity; the control of defects; functionalization with O, N, B or P heteroatoms; and the formation of graphene composites. These methodologies of structural design are needed for fast electrical charge storage/transfer and the transport of electrolyte ions (Li+, H+, K+, Na+) in graphene electrodes. We critically review state-of-the-art progress in the optimization of the electrochemical performance of graphene-based electrode materials. The structure of graphene needs to be designed to develop novel electrochemical energy storage devices that approach the theoretical charge limit of graphene and to deliver electrical energy rapidly and efficiently.
Co-reporter:Kunfeng Chen, Shu Yin and Dongfeng Xue
Nanoscale 2015 vol. 7(Issue 3) pp:1161-1166
Publication Date(Web):28 Nov 2014
DOI:10.1039/C4NR05880A
A new “combinatorial transition-metal cation pseudocapacitor” was demonstrated by designing combinatorial transition-metal cation pseudocapacitors with binary AxB1−x salt electrodes involving manganese, iron, cobalt, and nickel cations in an alkaline aqueous electrolyte. Binary multi-valence cations were crystallized in the colloidal state through an in situ coprecipitation under an electric field. These electroactive colloids absorbed by carbon black and the PVDF matrix are highly redox-reactive with high specific capacitance values, where the specific electrode configuration can create short ion diffusion paths to enable fast and reversible Faradaic reactions. This work shows huge promise for developing high-performance electrical energy storage systems via designing the colloidal state of electroactive cations. Multiple redox cations in the colloidal state can show high redox activities, making them more suitable for potential application in pseudocapacitor systems.
Co-reporter:Kunfeng Chen, Shuyan Song and Dongfeng Xue
Journal of Materials Chemistry A 2015 vol. 3(Issue 6) pp:2441-2453
Publication Date(Web):19 Dec 2014
DOI:10.1039/C4TA06989G
Much progress about graphene has been made in the fields of physics, chemistry, material science, and electronics. Graphene's properties are mainly dependent on its geometric structures and synthesis methods. Various newly developed chemical methods have been designed to tailor graphene materials with specific functionalities, such as crystallization routes, which can be a new direction in graphene R&D. In this review, we focus on recent developments in the synthesis of graphene materials with specific structures and electrochemical performances by top-down routes. First, ice crystallization from water molecules within graphene oxide is discussed to form 3D graphene oxide aerogel and graphene aerogel with porous networks. Then we review an in situ electrochemical crystallization route to fabricate graphene/metal oxide aerogel electrode materials. The electrochemical properties of different structural graphene types are discussed as lithium-ion batteries and supercapacitors. Future challenges and current progress beyond graphene as an energy storage material have been highlighted.
Co-reporter:Kunfeng Chen, Xu Chen and Dongfeng Xue
CrystEngComm 2015 vol. 17(Issue 9) pp:1906-1910
Publication Date(Web):29 Jan 2015
DOI:10.1039/C4CE02504K
In this work, we studied the crystallization of FeOOH nanorods via hydrolysis of FeCl3·6H2O solution under a low-temperature hydrothermal route (100 °C). The effect of Fe3+ concentration and solution pH on the crystallized morphology and size of FeOOH nanorods was systematically studied based on the chemical reaction and crystallization process. The electrochemical performance of the as-obtained FeOOH materials as supercapacitors is evaluated and the effect of Fe3+ concentration on the pseudocapacitance of iron-based materials is also discussed. FeOOH electrode materials obtained in 0.2 M FeCl3·6H2O solution display the highest specific capacitance of 714.8 F g−1, which is higher than reported values for FeOOH electrode materials. The present work demonstrates a simple hydrolysis route to synthesize high capacitance electrode materials.
Co-reporter:Keyan Li, Fenfen Shua, Xinwen Guo and Dongfeng Xue
CrystEngComm 2015 vol. 17(Issue 27) pp:5094-5100
Publication Date(Web):02 Jun 2015
DOI:10.1039/C5CE00811E
MnCO3 precursors with different morphologies were crystallized using three kinds of surfactants as soft templates, i.e., cation surfactant cetyl trimethyl ammonium bromide (CTAB), anion surfactant sodium dodecyl sulfate (SDS) and neutral poly(vinyl pyrrolidone) (PVP). When PVP was used, the reaction manner was changed from only stirring at room temperature to a hydrothermal route. Under hydrothermal conditions, different ethanol/water ratios and sources of CO32− (NaHCO3 and urea) were used. Porous cubic, regular spherical and nut-like spherical Mn2O3 samples can be obtained by a simple post-annealing process. The correlation between the morphology of Mn2O3 and its performance as an anode material for Li-ion batteries was evaluated. The nut-like spherical Mn2O3 sample has the best cycling performance, with a specific discharge capacity of 925 mA h g−1 at a current density of 100 mA g−1 after 180 cycles. The sample composed of cubes and spheres has superior rate performance. The specific discharge capacity decreases with increasing current density from ~872 mA h g−1 at 100 mA g−1 to ~361 mA h g−1 at 2000 mA g−1.
Co-reporter:Congting Sun and Dongfeng Xue
CrystEngComm 2015 vol. 17(Issue 13) pp:2728-2736
Publication Date(Web):02 Mar 2015
DOI:10.1039/C5CE00196J
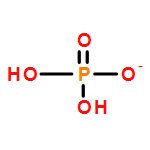
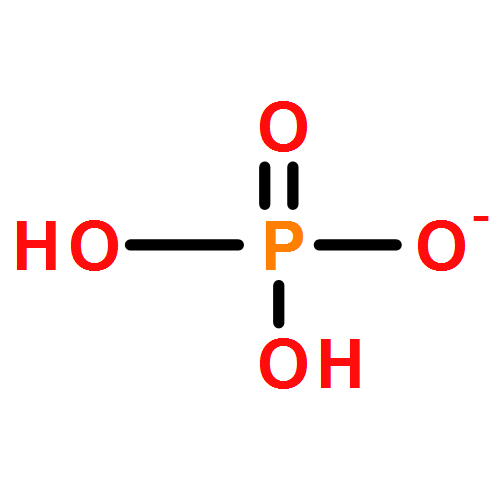
Both the crystallizing molecule without long-range ordered arrangement and the complex interactions between solvent, anti-solvent, and molecule crystal challenge the imaging of molecule configurations during anti-solvent crystallization. Here, the structural evolution of NH4H2PO4 during anti-solvent crystallization was tracked by in situ ATR-IR spectroscopy. The NH4H2PO4 molecule undergoes D2d → C2v → D2d or C2v → D2d during anti-solvent crystallization, depending on both V(H2O)/V(ethanol) ratio and the concentration of NH4H2PO4 aqueous solution. Moreover, IR spectroscopy can also be used to confirm the anti-solvent role of ethanol in the NH4H2PO4–H2O–ethanol crystallization system. The volatilization of ethanol has been demonstrated as an effective approach to control the dissolution process of NH4H2PO4, which can be used to study the crystallization–dissolution–recrystallization process. The present study can shed light on the anti-solvent crystallization physical and chemistry of NH4H2PO4 from a molecule-level viewpoint.
Co-reporter:Yan Wang, Congting Sun, Chaoyang Tu and Dongfeng Xue
CrystEngComm 2015 vol. 17(Issue 15) pp:2929-2934
Publication Date(Web):04 Mar 2015
DOI:10.1039/C5CE00291E
The chemical bonding theory of single crystal growth was applied to a Gd3Ga5O12 (GGG) Czochralski growth system. On the basis of anisotropic chemical bonding characteristics, the mesoscale process controlled by both thermodynamics and kinetics has been studied in GGG single crystal growth. Starting from the unit-scale in the growth system, the mesoscale structures undergo evolution from GdO8, GaO6, and GaO4 to Gd3Ga5O12 clusters and to a GGG single crystal via chemical bonding between Gd, Ga, and O. On the basis of the chemical bonding theory of single crystals, the GGG single crystal thermodynamically prefers to exhibit a hexagonal configuration along the [111] pulling direction. Kinetically, isotropic mass transfer in the Czochralski growth process leads to formation of a GGG single crystal with a circular configuration, as viewed down along the [111] direction. In such a case, the GGG single crystal maintains the lowest energy. In order to satisfy both thermodynamic and kinetic controls, the thermodynamically preferred {−110} microfacets are exposed on the microscale and the single crystal adopts a circular shape on the macroscale. The present work deepens our understanding of the mesoscale process in the GGG Czochralski growth system.
Co-reporter:Kunfeng Chen, Congting Sun and Dongfeng Xue
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 2) pp:732-750
Publication Date(Web):10 Nov 2014
DOI:10.1039/C4CP03888F
Advances in materials have preceded almost every major technological leap since the beginning of civilization. On the nanoscale and microscale, mastery over the morphology, size, and structure of a material enables control of its properties and enhancement of its usefulness for a given application, such as energy storage. In this review paper, our aim is to present a review of morphology engineering of high performance oxide electrode materials for electrochemical energy storage. We begin with the chemical bonding theory of single crystal growth to direct the growth of morphology-controllable materials. We then focus on the growth of various morphologies of binary oxides and their electrochemical performances for lithium ion batteries and supercapacitors. The morphology–performance relationships are elaborated by selecting examples in which there is already reasonable understanding for this relationship. Based on these comprehensive analyses, we proposed colloidal supercapacitor systems beyond morphology control on the basis of system- and ion-level design. We conclude this article with personal perspectives on the directions toward which future research in this field might take.
Co-reporter:Congting Sun and Dongfeng Xue
Crystal Growth & Design 2015 Volume 15(Issue 6) pp:2867
Publication Date(Web):May 12, 2015
DOI:10.1021/acs.cgd.5b00293
Unravelling the mesoscale process and the dynamic heterogeneous structures that appear on the mesoscale in a crystallization system is important in designing and fabricating functional crystalline materials. Recent experimental observations show the existence of stable clusters and amorphous intermediates before the formation of a crystalline solid, which seems to contradict classical nucleation theory. Here we show by in situ infrared spectroscopy and theoretical calculation that the liquid/solid phase transformation of urea proceeds through the agglomeration of primary clusters. The phase transformation pathway of urea in solution has been identified, in which urea molecules initially aggregate into one-dimensional (1D) molecular chains, and then these 1D molecular chains assemble to 2D plane-like and 3D net-like clusters. Crystalline urea with P–421m symmetry can be formed when these 3D net-like clusters overcome a critical size. Both experimental and calculated results demonstrate that the liquid/solid phase transformation of urea in aqueous solution obeys the classical nucleation theory. Finally, a morphology diagram of urea is provided on the basis of relative chemical bonding energy density. This morphology diagram can be used to understand the multiple anisotropic geometries for how urea crystals in an aqueous solution system can be laid out. Our results demonstrate the concept of identifying a particular mesoscale process in a urea crystallization system by both in situ vibration spectroscopy observations and chemical bonding calculations.
Co-reporter:Xu Chen, Kunfeng Chen, Hao Wang, Dongfeng Xue
Journal of Colloid and Interface Science 2015 Volume 444() pp:49-57
Publication Date(Web):15 April 2015
DOI:10.1016/j.jcis.2014.12.026
An electrochemical system including functioned Fe(NO3)3 and alkaline electrolyte is constructed to study the charge storage mechanism upon both thermodynamic calculations and electrochemical measurements. The thermodynamic calculation results demonstrate that increasing KOH concentration of alkaline electrolyte can enhance electrolyte activity but decrease the peak potential, which agrees well with that of electrochemical measurement. The present results indicate that the proposed pseudocapacitive redox reactions are between Fe3+ and Fe2+ in our salt electrode of Fe(NO3)3 system, in which the solid Fe3+ in FeOOH colloids serves as the active anode component and free Fe3+ on the colloidal electrodes serves as the cathode component. The active central ions of Fe3+ in Fe(NO3)3⋅9H2O can be fixed on the electrodes by the surrounding ligands (OH− and NO3−) and can be in-situ transformed into colloidal Fe4NO3(OH)11 and goethite (α-FeOOH). Electrochemical results indicate that the current proposed colloidal pseudocapacitor system warrants the high-efficiency utilization of electroactive central Fe3+ ions, showing high energy density of 58.4 Wh/kg at the power density of 8.4 kW/kg as an anode material. Meanwhile, our designed pseudocapacitor system can function well as a supercapacitor cathode. This colloidal pseudocapacitor system can offer a facile and efficient route for the design of advanced supercapacitors.
Co-reporter:Kunfeng Chen, Shuyan Song and Dongfeng Xue
CrystEngComm 2015 vol. 17(Issue 10) pp:2110-2117
Publication Date(Web):21 Jan 2015
DOI:10.1039/C4CE02340D
Micro- and nanocrystals with different facets may exhibit different chemical activities that are of great importance in practical applications. The electrochemical performances of Cu2O anodes with different crystal planes are compared. This work comprehensively investigates the effect of the crystal plane on the electrochemical performance of Cu2O. Firstly, Cu2O polyhedra with different morphologies, i.e., cube, octahedron and truncated octahedron, with different degrees of exposed {110} facets, were synthesized. Cu2O cubes show the highest capacity among these Cu2O polyhedra. The present results prove that the {100} facets of Cu2O display high electroactivities toward redox reactions. The electroactivity sequence of the crystal planes is {100} > {111} > {110}. These results confirm that the additional capacity of Cu2O is due to the generation of CuO, which can conduct a two-electron redox reaction. The anode performances of Cu2O polyhedra are dependent on planes, and this can provide a novel route to design high performance electrode materials for lithium-ion batteries.
Co-reporter:Congting Sun, Yan Wang, Chaoyang Tu and Dongfeng Xue
CrystEngComm 2015 vol. 17(Issue 17) pp:3208-3213
Publication Date(Web):20 Mar 2015
DOI:10.1039/C5CE00339C
In the flux system of GdAl3(BO3)4, the compromise in competition between thermodynamic and kinetic control on the mass transfer has been studied from the viewpoint of chemical engineering. Different control mechanisms on the mass transfer in the melt growth system result in various mesoscale morphologies of a single crystal. Both the calculated and experimental results demonstrate thermodynamic control of the anisotropic mass transfer during crystal growth, leading to the mesoscale morphology evolution of the GdAl3(BO3)4 single crystal similar to the matryoshka. The present work can deepen our understanding of mesoscale morphology evolution of the GdAl3(BO3)4 single crystal in the flux growth system.
Co-reporter:Keyan Li, Fenfen Shua, Jiawei Zhang, Kunfeng Chen, Dongfeng Xue, Xinwen Guo, Sridhar Komarneni
Ceramics International 2015 Volume 41(Issue 5) pp:6729-6733
Publication Date(Web):June 2015
DOI:10.1016/j.ceramint.2015.01.116
Orthorhombic LiMnO2 (o-LiMnO2) was prepared under hydrothermal conditions using sol–gel derived Mn2O3 and LiOH as precursors and the effects of hydrothermal reaction parameters such as reaction temperature and LiOH and Mn2O3 concentrations on the phase purity of o-LiMnO2 were studied. The as-prepared samples were characterized by X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), and galvanostatic charge−discharge tests. Phase purity of o-LiMnO2 was affected by hydrothermal temperature and LiOH and Mn2O3 concentrations. High LiOH concentration facilitated the synthesis of pure phase o-LiMnO2. Low reaction temperature, lower than 140 °C, did not lead to the formation of pure phase of o-LiMnO2. Galvanostatic charge–discharge test results showed that pure o-LiMnO2 had larger discharge capacity than the mixed phases of o-LiMnO2 and Li2MnO3. However, the cycling performance of the mixed phases of o-LiMnO2 with Li2MnO3 was found to be better than that of pure o-LiMnO2.
Co-reporter:Fei Liu
Science China Technological Sciences 2015 Volume 58( Issue 11) pp:1841-1850
Publication Date(Web):2015 November
DOI:10.1007/s11431-015-5932-y
Graphene is a promising material as both active components and additives in electrochemical energy storage devices. The properties of graphene strongly depend on the fabrication methods. The applications of reduced graphene oxide as electrode materials have been well studied and reviewed, but the using of “pristine” graphene as electrode material for energy storage is still a new topic. In this paper, we review state-of-the-art progress in the fabrication of “pristine” graphene by different methods and the electrochemical performance of graphene-based electrodes. The achievements in this area will be summarized and compared with the graphene oxide route in terms of cost, scalability, material properties and performances, and the challenges in these methods will be discussed as well.
Co-reporter:KunFeng Chen
Science China Technological Sciences 2015 Volume 58( Issue 11) pp:1768-1778
Publication Date(Web):2015 November
DOI:10.1007/s11431-015-5915-z
The search of electrode materials with high electrochemical activity is one of key solutions to actualize both high energy density and high power density in a supercapacitor. Recently, we have developed one novel kind of rare earth and transitional metal colloidal supercapacitors, which can deliver higher specific capacitance than electrical double-layer capacitors (EDLC) and traditional pseudocapacitors. The electrode materials in colloidal supercapacitors arin colloidal supercapacitors aree in-situ formed electroactive colloids, which were transformed from commercial rare earth and transitional metal salts in alkaline electrolyte by chemical and electrochemical assisted coprecipitation. In these colloidal supercapacitors, multiple-electron Faradaic redox reactions can be utilized, which can deliver ultrahigh specific capacitance often larger than one-electron capacitance. Multiple-valence metal cations used in our designed colloidal supercapacitors mainly include Ce3+, Yb3+, Er3+, Fe3+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Sn2+ and Sn4+. The colloidal supercapacitors can be served as the promising next-generation high performance supercapacitors.
Co-reporter:Kunfeng Chen, Yangyang Yang, Keyan Li, Zengsheng Ma, Yichun Zhou, and Dongfeng Xue
ACS Sustainable Chemistry & Engineering 2014 Volume 2(Issue 3) pp:440
Publication Date(Web):November 6, 2013
DOI:10.1021/sc400338c
The major limitation of supercapacitors is their low energy densities compared with battery systems. However, there has not been an advanced charge storage mechanism for increasing the electrochemical performance of pseudocapacitors. For the first time, we reported that water-soluble CoCl2 electrodes can show a reversible redox reaction of Co2+ ↔ Co3+ ↔ Co4+ on the electrode and deliver very high specific pseudocapacitance of ∼1962 F/g. Commercial CoCl2 salt was used directly as pseudocapacitor electrodes in an aqueous electrolyte neglecting the complex synthesis procedures. We further provided electronegativity as a theoretical guideline to identify the promising active metal cations in our pseudocapacitor system. The new charge storage mechanism based on active cations offers critical insights to the rational design of a new generation of energy storage devices.Keywords: Active metal cation; CoCl2; Inorganic salt; Pseudocapacitor; Supercapacitor; Ultrahigh specific capacitance
Co-reporter:Xu Chen, Kunfeng Chen, Hao Wang, Dongfeng Xue
Electrochimica Acta 2014 Volume 147() pp:216-224
Publication Date(Web):20 November 2014
DOI:10.1016/j.electacta.2014.08.132
For the first time, we designed a Fe(NO3)3 salt supercapacitor in alkaline aqueous electrolyte, which can be served as active components of both positive and negative electrodes and has high potential window of 1.6 V, higher than the theoretical operating voltage of water (1.23 V). After undergoing the chemical coprecipitation and electrochemical redox reactions in KOH electrolyte, Fe3+ cations were in-situ crystallized into high electrochemical active goethite FeOOH colloids. The charge storage mechanism observed in both positive and negative electrodes is pseudocapacitive reaction between FeOOH and Fe(OH)2 phases. In the three-electrode system, Fe(NO3)3 electrodes showed wider negative potential window (−1.2−0 V) compared to positive potential window (0−0.45 V). The Fe(NO3)3 negative electrodes exhibited 120 F/g capacitance, 23.6 Wh/kg energy density and 0.6 kW/kg power density, while the Fe(NO3)3 positive electrodes displayed 393 F/g, 10.4 Wh/kg and 0.22 kW/kg at the current density of 1 A/g. As positive and negative electrodes, Fe cation utilization ratios can reach 72% and 60%, respectively. The assembled symmetric Fe(NO3)3 supercapacitor device can exhibit remarkable energy density of 11.9 Wh/kg at the power density of 0.4 kW/kg. For pure iron oxides/hydroxides electrodes, these values are larger than that reported in previous literatures. The present results demonstrated the high performances of Fe(NO3)3 symmetric supercapacitors and the potentialities of using Fe(NO3)3 electrodes as negative and/or positive electrodes for supercapacitors with high energy densities.
Co-reporter:Keyan Li, Hao Chen, Fenfen Shua, Kunfeng Chen, Dongfeng Xue
Electrochimica Acta 2014 Volume 136() pp:10-18
Publication Date(Web):1 August 2014
DOI:10.1016/j.electacta.2014.05.068
Amorphous FeOOH and α-FeOOH were synthesized by simple solvothermal methods and used as the reactants to prepare iron oxide-based cathode materials. Amorphous FeOOH transforms to α-Fe2O3 at temperatures as low as 80 °C, and is stable enough to avoid the transformation to α-LiFeO2 even at 200 °C in LiOH ethanol solution under solvothermal condition. α-FeOOH transforms to α-LiFeO2 in the same reaction system at 140 °C. Both α-Fe2O3 and α-LiFeO2 demonstrate good electrochemical performances, especially α-Fe2O3, which delivers a high reversible capacity (105 mAh/g at 100 mA/g after 50 cycles) in the voltage range from 1.5 to 4.5 V. The electrochemical performances of α-Fe2O3 were found to be influenced by the solvent, temperature, reaction duration and alkali concentration of the solvothermal reactions. It was found that ethanol solution, low temperature, and moderate reaction duration and alkali concentration lead to α-Fe2O3 with reversible capacity of about 100 mAh/g.
Co-reporter:Kunfeng Chen, Fei Liu, Shuyan Song and Dongfeng Xue
CrystEngComm 2014 vol. 16(Issue 33) pp:7771-7776
Publication Date(Web):24 Jun 2014
DOI:10.1039/C4CE01030B
We proved that ice crystallized from water molecules within as-synthesized graphene oxide (GO) can be used as a spacer among individual GO sheets to form an expanded GO aerogel with a three-dimensional porous network structure. This designed specific structure can be retained during further processes to form graphene paper. Due to the fact that the as-fabricated graphene paper contains carbon defects and space in its infrastructure, this creates more electroactive surfaces and interfaces to significantly increase the Faradaic reaction efficiency. With the introduction of redox-electrolyte K3Fe(CN)6 into a KOH electrolyte, we can obtain a 95-fold increase in the specific capacitance compared with the value obtained in the traditional KOH electrolyte. The ice-crystallization formed graphene paper electrode showed high electroactivity because the specific porous structure can favor the fast transfer of electrolyte ions, such as Fe(CN)63− ions. The presented results prove that ice-crystallization formed GO can serve as a chemically tunable platform for electrochemical energy storage applications.
Co-reporter:Xu Chen, Kunfeng Chen, Hao Wang, Shuyan Song and Dongfeng Xue
CrystEngComm 2014 vol. 16(Issue 29) pp:6707-6715
Publication Date(Web):25 Apr 2014
DOI:10.1039/C4CE00660G
For the first time, we have studied the crystallization behavior of FeCl3 in an aqueous alkaline pseudocapacitor system, where FeCl3 can be effectively transformed into Fe2O3·H2O colloids by an in situ crystallization process in KOH electrolyte under an electric field. In alkaline aqueous solution, the Fe3+ cations firstly react with OH− and crystallize into goethite Fe2O3·H2O colloids. During the electrochemical tests, the crystallization of Fe3+ was interrupted by an external electric field due to a Faradaic redox reaction, resulting in fine and highly electroactive Fe2O3·H2O colloids. For the currently designed FeCl3 pseudocapacitor system, a very high capacitance of 977 F g−1 at the current density of 3 A g−1 and a high energy density of 23.6 Wh kg−1 at the power density of 3400 W kg−1 can be obtained. Rate performance tests show that 86% of the capacitance can be maintained after the galvanostatic current density increases from 3 to 15 A g−1. Our current pseudocapacitor system can provide a versatile method for the construction of high-performance inorganic pseudocapacitors by in situ crystallization via chemical/electrochemical reactions in room temperature aqueous solution.
Co-reporter:Keyan Li, Shudong Lin, Fenfen Shua, Jiawei Zhang, Kunfeng Chen and Dongfeng Xue
CrystEngComm 2014 vol. 16(Issue 48) pp:10969-10976
Publication Date(Web):13 Oct 2014
DOI:10.1039/C4CE01882F
Nanocrystalline spinel LiMn2O4 and layered LiCo1−xMnxO2 (x = 0–0.15) cathode materials were synthesized by a rapid combustion route in combination with an annealing treatment using common filter paper as the template and ethanol as the fuel. The structure, morphology and electrochemical properties of the materials were studied by X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), galvanostatic charge–discharge test, cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). The spinel LiMn2O4 annealed at 750 °C shows excellent cycling stability (capacity retention is 92% after 200 cycles), high coulombic efficiency (>99%) and good rate capability. The layered LiCoO2 shows high initial capacity (168.9 mAh g−1) and good rate capability, but its capacity retention is only 74% after 80 cycles owing to the nanosize effect. After doping a small quantity of Mn (x = 0.05), the cycling performance of the Mn-doped sample was significantly improved compared with that of the pristine LiCoO2 (capacity retention is 87% after 80 cycles).
Co-reporter:Jingwen Zhu and Dongfeng Xue
CrystEngComm 2014 vol. 16(Issue 4) pp:642-648
Publication Date(Web):21 Oct 2013
DOI:10.1039/C3CE41905C
We report an electrochemically modulated method for single crystal silver nanobelts with uniform thickness and width, which grow along the <110> direction with {111} facets. The procedure involves a crystallographically-oriented and interfacial kinetic controlled chemical reduction process by using a sacrificial cathode electrode with nanochannel templates. Silver nanobelt-bundle arrays are formed orientedly within nanochannels and can be easily dispersed into individual nanobelts by removing the template. These nanobelts exhibit two distinct surface plasmon resonances (SPR) at 456 and 980 nm, and show a strong spontaneous emission (SE) at 519 nm when excited by UV or blue light. These silver nanobelts with a quasi-two-dimensional planar nanostructure are an important material for surface plasmon resonance-based technology, which may open up many practical opportunities in the fields of optics, electronics and photoelectronics.
Co-reporter:Kunfeng Chen and Dongfeng Xue
CrystEngComm 2014 vol. 16(Issue 21) pp:4610-4618
Publication Date(Web):20 Mar 2014
DOI:10.1039/C4CE00380B
In the present work, we designed an in situ crystallization method for tin chloride salts pseudocapacitors, that is, a SnCl4·5H2O or SnCl2·2H2O electrode in aqueous KOH electrolyte. After undergoing coupled chemical/electrochemical crystallization and Faradaic redox reactions, highly active SnO/Sn colloids were in situ crystallized within the carbon black matrix; such an electrode configuration can result in the fast transfer of ions/electrons and efficiently utilize the active tin cations in the salt electrode, thus high specific capacitance can be obtained. The SnCl4 electrode can deliver ultrahigh specific capacitance of 1592 F g−1 at a current density of 1 A g−1 and a potential range of 0.42 V, which is the highest value among the reported tin-based supercapacitors. Our designed tin chloride electrode/alkaline electrolyte system can offer critical insights into the rational design of the next generation of high-performance supercapacitor devices.
Co-reporter:Congting Sun and Dongfeng Xue
CrystEngComm 2014 vol. 16(Issue 11) pp:2129-2135
Publication Date(Web):28 Nov 2013
DOI:10.1039/C3CE42292E
The growth of YAG bulk crystals was studied using both theoretical calculations based on the anisotropic chemical bonding conditions and practical growth via the Czochralski (Cz) method. The chemical bonding theory of single crystal growth quantitatively describes the anisotropic bonding behaviors of constituent atoms during crystallizing, which can be applied to the thermodynamic growth of YAG single crystals. Both bonding conditions and crystal symmetry determine the projection configuration along the pulling direction and crystal ridges in the crystal shoulder of YAG grown along [111] direction. During Cz growth process of YAG single crystals, the relative low growth rate along <110> directions results in the exposure of surfaces normal to <110> directions. However, the chemical bonding energy density at the intersection of two adjacent <110> growth directions is higher, leading to the exposure of surfaces normal to <112> directions and the truncated-hexagon configuration of YAG along [111] direction. ϕ 3′′ YAG single crystal was successfully grown. Our present work provides a promising approach to achieve controllable growth for functional bulk crystal via both thermodynamic and kinetic controls.
Co-reporter:Kunfeng Chen, Congting Sun, Shuyan Song and Dongfeng Xue
CrystEngComm 2014 vol. 16(Issue 24) pp:5257-5267
Publication Date(Web):06 Mar 2014
DOI:10.1039/C4CE00339J
Cu2O can crystallize into various polymorphs, such as cubes, rhombic dodecahedra, branching structures, and hopper cubes, instead of thermodynamically stable octahedra by designed kinetic-control routes instead of traditional thermodynamic control. The present results confirmed that Cu2O polymorphs have distinct physical and chemical properties, and morphology changes can occur due to polymorphic transitions in a kinetics-controllable reaction system. Both thermodynamic and kinetic factors on these polymorphism systems are believed to be significant for developing a polymorphism–property relationship, ultimately guiding the appropriate material selection for specific applications. Furthermore, we took Cu2O as an example to illustrate the development of “polymorphism” in modern materials science: polymorphism is an intrinsic physicochemical characteristic, and involves varying growth shapes, phase transformation and variation of physical properties. These findings can provide new insight to polymorphism–performance correlation and kinetic–thermodynamic control synthesis.
Co-reporter:Kunfeng Chen and Dongfeng Xue
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 23) pp:11168-11172
Publication Date(Web):12 Mar 2014
DOI:10.1039/C4CP00811A
An ex situ observation was made by XRD patterns, SEM and TEM images, as well as cyclic voltammogram curves of CuO/Cu integrated anodes for lithium ion batteries. For the first time, the existence of a Cu+ ion long-range transfer path was identified at the potential widow of 1.30–1.60 V during both charging and discharging processes. Both SEM and TEM images show that these nanowires networks hanging CuO nanoparticles provide a Cu+ diffusion path within our designed CuO/Cu integrated anode. This work provides new insights into the conversion reaction of inorganic anode materials, and can favor the development of high-performance conversion anodes for lithium-ion batteries.
Co-reporter:Congting Sun and Dongfeng Xue
Crystal Growth & Design 2014 Volume 14(Issue 5) pp:2282-2287
Publication Date(Web):April 11, 2014
DOI:10.1021/cg401867c
The anisotropic growth of large-size sapphire single crystals along different pulling directions was studied on the basis of the chemical bonding theory of single crystal growth and practical Czochralski growth. The projection of thermodynamic morphology of sapphire single crystal respectively along [210], [110], [001], and [001] rotated 57.62° directions can be used to confirm the growth directions of surfaces that are preferred to be exposed thermodynamically in Czochralski growth. Starting from these thermodynamically preferred directions, the possible radial directions that are normal to the four typical pulling directions by kinetic controls have been identified by anisotropic chemical bonding distributions of sapphire single crystal. Chemical bonding calculations demonstrate that the lower pulling rate should be designed when Raxial/Rradial > 1, whereas the higher pulling rate should be designed when Raxial/Rradial < 1. The anisotropic chemical bonding conditions demonstrate the lowest chemical bonding density along the radial directions of sapphire single crystal when it grows along the [001] pulling direction. Taking [001] as the pulling direction in practical growth, a ϕ 2″ sapphire single crystal was grown via the Czochralski method with a growth rate of 2–3 mm/h. Our present work shows the effect of anisotropy on the Czochralski growth of large-size single crystals, which can provide a theoretical guide in practical growth from both thermodynamic and kinetic viewpoints.
Co-reporter:Keyan Li, Hao Chen, Fenfen Shua, Dongfeng Xue and Xinwen Guo
RSC Advances 2014 vol. 4(Issue 69) pp:36507-36512
Publication Date(Web):14 Aug 2014
DOI:10.1039/C4RA06889K
Iron oxide@C composites and lithium ferrites were synthesized by a cotton-template method as anode materials for Li-ion batteries. α-Fe2O3@C composites with 3-D porous hollow secondary structures were prepared by directly burning the cotton containing the iron salt (FeCl3 or Fe(NO3)3) in air, and the Fe3O4@C composites with similar structures were obtained by annealing α-Fe2O3@C under a N2 atmosphere. α-LiFeO2 and α-LiFe5O8 particles with sizes of 100–500 nm were also prepared using a similar method. Electrochemical measurements showed that all these samples demonstrated good electrochemical performances as Li-ion battery anodes, especially α-Fe2O3@C derived from Fe(NO3)3, which delivers a high reversible capacity of 990 mA h g−1 at 100 mA g−1 after 50 cycles. Both the porous hollow secondary structure and the suitable amount of amorphous carbon are significant for the electrochemical performances of the iron oxide@C composites. Such a method is simple, rapid and inexpensive and may facilitate the preparation of other high performance electrode materials with porous hollow structures.
Co-reporter:Kunfeng Chen, Young Dong Noh, Wenyan Huang, Jianfeng Ma, Sridhar Komarneni, Dongfeng Xue
Ceramics International 2014 Volume 40(Issue 2) pp:2877-2884
Publication Date(Web):March 2014
DOI:10.1016/j.ceramint.2013.10.024
Fe-based materials, Fe2O3, Fe3O4, and FeOOH, were synthesized by the microwave–hydrothermal process in the temperature range of 100–200 °C and under very short reaction times of 15 min to 2 h. Under microwave-controlled hydrolysis and redox reactions, cube-like Fe2O3 was crystallized using FeCl3, Fe3O4 particles were crystallized from FeCl2 and FeOOH nanorods were crystallized using FeCl3. The Fe-based materials were fabricated to make anodes and cathodes of lithium-ion battery and supercapacitor electrode materials to study their potential electrochemical applications. The electrochemical results showed that FeOOH had better anode capacity as lithium-ion batteries than those of Fe2O3 and Fe3O4. The present results suggest that the microwave–hydrothermally synthesized Fe-based materials are promising lithium-ion battery anode materials.
Co-reporter:Kunfeng Chen, Shuyan Song and Dongfeng Xue
RSC Advances 2014 vol. 4(Issue 44) pp:23338-23343
Publication Date(Web):05 May 2014
DOI:10.1039/C4RA03037K
We designed a new type of ionic pseudocapacitor system with excellent contributions from ionic-state redox mediators, including a redox couple [Fe(CN)6]3−/[Fe(CN)6]4− in the electrolyte and redox cations in highly electroactive colloid electrodes. The highest pseudocapacitance value of 12658 mF cm−2 was obtained, which is a 7-fold increase in the specific capacitance of the CoCl2 electrode for K3Fe(CN)6 in a KOH alkaline electrolyte at a current density of 20 mA cm−2 and at a potential interval of 0.55 V. The currently designed supercapacitor systems show a versatile strategy to design high-performance supercapacitors with CoCl2, CuCl2, NiCl2, and FeCl3 electrodes.
Co-reporter:Kunfeng Chen, Ailaura C. Donahoe, Young Dong Noh, Keyan Li, Sridhar Komarneni, Dongfeng Xue
Ceramics International 2014 Volume 40(Issue 2) pp:3155-3163
Publication Date(Web):March 2014
DOI:10.1016/j.ceramint.2013.09.128
Abstract
The LiMn2O4 electrode materials were synthesized by the conventional-hydrothermal and microwave-hydrothermal methods. The electrochemical performances of LiMn2O4 were studied as supercapacitors in LiNO3 electrolyte and lithium-ion battery cathodes. The microwave-hydrothermal method can synthesize LiMn2O4 electrode materials with reversible electrochemical reaction in a short reaction time and low reaction temperature than conventional-hydrothermal route. The capacitance of LiMn2O4 electrode increased with increasing crystallization time in conventional-hydrothermal route. The results showed that LiMn2O4 supercapacitors had similar discharge capacity and potential window (1.2 V) as that of ordinary lithium-ion battery cathodes. In LiNO3 aqueous electrolyte, the reaction kinetics of LiMn2O4 supercapacitors was very fast. Even, at current densities of 1 A/g and 5 A/g, aqueous electrolyte gave good capacity compared with that in organic electrolyte at a current density of 0.05 A/g.
Co-reporter:Kunfeng Chen, Young Dong Noh, Rinkal R. Patel, Wenyan Huang, Jianfeng Ma, Keyan Li, Sridhar Komarneni, Dongfeng Xue
Ceramics International 2014 Volume 40(Issue 6) pp:8183-8188
Publication Date(Web):July 2014
DOI:10.1016/j.ceramint.2014.01.014
Crystalline Co3O4 and Co(OH)2 were synthesized using Co(NO3)2 as a precursor by conventional–hydrothermal and microwave–hydrothermal routes, respectively. The Co3O4 phase showed cubic morphologies while the β-Co(OH)2 phase exhibited plate-like shapes. The electrochemical performances of Co3O4 and Co(OH)2 phases were evaluated as electrode materials for lithium-ion battery anodes, cathodes and supercapacitors. Both Co3O4 and Co(OH)2 phases showed pseudocapacitive performances in Li2SO4 and KOH electrolytes. The Co3O4 and Co(OH)2 phases were found to be more promising as anodes than as cathodes in lithium-ion batteries. The Co(OH)2 electrodes showed higher specific capacitances than those of Co3O4 materials.
Co-reporter:Congting Sun, Dongfeng Xue
Journal of Molecular Structure 2014 Volume 1059() pp:338-342
Publication Date(Web):5 February 2014
DOI:10.1016/j.molstruc.2013.11.042
•The hydrogen bonding nature during ADP crystallization is studied.•The crystallographic structures of NH4+ and H2PO4- in ADP crystal are discussed.•The hydrogen bonding of NH4+ and H2PO4- is recorded by in situ IR spectrum.•Anisotropic chemical bonding conditions dominate ADP crystal morphology.•ADP crystal morphologies with different H2PO4-n clusters are calculated.The hydrogen bonding nature during ADP crystallization is studied on the basis of anisotropic chemical bonding conditions in ADP crystal combined with in situ IR observation. The variations of hydrogen bonding nature of NH4+ and H2PO4- groups dominate the transformation from the free hydrated ionic state to crystalline state during ADP crystallization. Anisotropic ADP crystal morphology depends on the anisotropic chemical bonding conditions along [1 0 0] and [1 0 1] directions. ADP crystal morphologies with different H2PO4-n (n = 1–8) clusters can be calculated on the basis of hydrogen bonding conditions and H2PO4-n cluster structures at the growth interface. Experimentally, in situ IR spectrum can record the breaking of P–O⋯H–O–H and H–N⋯H–O–H, and the formation of P–O⋯H–O–P and H–N⋯H–O–P hydrogen bonding during ADP crystallization. The present work provides a promising strategy to identify the chemical bonding nature during crystallization processes of molecular crystals from aqueous solution.Graphical abstract
Co-reporter:Congting Sun, Dongfeng Xue
Optical Materials 2014 Volume 36(Issue 12) pp:1966-1969
Publication Date(Web):October 2014
DOI:10.1016/j.optmat.2013.12.019
•The chemical bonding theory of single crystal growth was applied to KDP and ADP.•The chemical bonding of K+/NH4+ and (H2PO4-)n framework dominates growth morphology.•〈1 0 0〉 and 〈1 0 1〉 are possible preferential growth directions for the dendrite growth.•IR spectroscopy of KDP and ADP solutions testified our calculations and assumptions.•H2PO4- ions initially form (H2PO4-)n framework, cations then inset into this frame.Crystallographically, KDP and ADP have familiar (H2PO4-)n framework, which is formed via hydrogen bonding between H2PO4- groups along main crystallographic axes. The common characteristics of both KDP and ADP crystallization behaviours are studied from the viewpoint of (H2PO4-)n framework. On the basis of chemical bonding theory of single crystal growth, the anisotropic hydrogen bonding in (H2PO4-)n framework and the chemical bonding between K+/NH4+ and (H2PO4-)n dominate the anisotropic thermodynamic equilibrium morphology of KDP and ADP. The thermodynamic equilibrium morphology is used to find out the possible preferential growth directions for the dendrite growth of both KDP and ADP, i.e., 〈1 0 0〉 and 〈1 0 1〉 directions. IR spectroscopy of both KDP and ADP solutions with different concentrations has testified our theoretical results, which shows that H2PO4- ions are initially form (H2PO4-)n framework, and then constituent cations inset into the (H2PO4-)n framework during both KDP and ADP crystallization processes. The present work can provide helpful basic information for the study of crystallization of MH2XO4 crystal family.Graphical abstract
Co-reporter:Congting Sun
The Journal of Physical Chemistry C 2014 Volume 118(Issue 29) pp:16043-16050
Publication Date(Web):July 3, 2014
DOI:10.1021/jp504830u
The competing occupancy of cation position by NH4+ and K+ during crystallization produces local distortions in structure, resulting in the difficulties in growing high-quality (K,NH4)H2PO4 single crystals with particular mixing concentration. Crystal properties such as structure, defect density, and purity always depend on the early formation stage of crystalline (K,NH4)H2PO4 molecule. Identification of the effect of K+/NH4+ mole ratios in KH2PO4–NH4H2PO4 aqueous solution on (K,NH4)H2PO4 molecule motion at the early stage of crystallization can provide the strategy to growing high-quality (K,NH4)H2PO4 crystals with particular mixing concentration. In situ molecule vibration spectroscopy was used to identify the early formation stage of crystalline (K,NH4)H2PO4 in KH2PO4–NH4H2PO4 aqueous solution with various K+/NH4+ mole ratios. (K,NH4)H2PO4 molecule motion was imaged via integrating the structural information on IR/Raman-active NH4+, H2PO4–, and hydrogen bonding infrastructures. K+/NH4+ mole ratio in KH2PO4–NH4H2PO4 aqueous solution determines the supersaturation in crystallization system as well as the competing incorporation of K+ and NH4+ into the lattice. Both lower supersaturation and stronger competition between cations hinder the crystallization of (K,NH4)H2PO4, resulting in the remarkable spectral difference before and after the formation of crystalline (K,NH4)H2PO4. Our results demonstrate the concept of competed incorporation between different cations into the anionic framework from the molecular viewpoint.
Co-reporter:Keyan Li, Congying Kang, and Dongfeng Xue
Inorganic Chemistry 2013 Volume 52(Issue 17) pp:10206-10210
Publication Date(Web):August 22, 2013
DOI:10.1021/ic401805x
We proposed a simple and an effective method to predict the site occupancy and threshold concentration of metal ions in lithium niobate (LiNbO3, LN) single crystal. The ionic energy parameter Ei, defined by the ionic electronegativity and ionic radius, was proposed to describe the electrostatic and size effects of cations on the structural stability of LN. The dopant location can be easily identified by comparing the Ei deviation of dopant from those of host cations Li+ and Nb5+, and the dopant prefers to occupy the lattice site with the smaller deviation of Ei. Our calculated occupancies agree well with those experimental results, which demonstrate the predictive power of our present method. We in this work predicted the preferred occupancies of 60 metal ions in LN single crystal. Further, the threshold concentrations of some frequently used dopants were calculated on the basis of the assumption that all doped LN crystals can endure the same variation of Ei.
Co-reporter:Kunfeng Chen, Shuyan Song and Dongfeng Xue
CrystEngComm 2013 vol. 15(Issue 46) pp:10028-10033
Publication Date(Web):09 Oct 2013
DOI:10.1039/C3CE41745J
Uniform hollow octahedra of Cu2O were successfully synthesized by the facile room-temperature chemical reaction of copper acetate and N2H4 without using any surfactant. The growth mechanism of the hollow octahedra and the solution environment in the chemical reaction were investigated. The chemical reactions from Cu2+ to Cu(OH)42− and finally to Cu2O can be done within <2 s. In this reaction system, N2H4 served as an alkali and reducing agent. Acetate ions, Ac−, can serve as buffer, act as counterions, and Ac− are coordinated to the Cu2O particle surface. The hollow-octahedron@nanoparticles-aggregate core@shell structures were synthesized by the introduction of NH4+ cations. The core@shell structures show a higher capacity and better cycling stability than the hollow octahedra as lithium-ion battery anodes because the core@shell structures can endure large volume changes during electrochemical reactions and nanoparticles with small sizes can shorten the diffusion path of the Li+ ions and electrons. As supercapacitors, the core@shell structures have a higher specific capacitance than that of the hollow octahedra.
Co-reporter:Kunfeng Chen, Shuyan Song, Keyan Li and Dongfeng Xue
CrystEngComm 2013 vol. 15(Issue 47) pp:10367-10373
Publication Date(Web):27 Sep 2013
DOI:10.1039/C3CE41802B
Currently, one of the biggest challenges in the field of pseudocapacitors is their capacitance value. Current knowledge on improving specific capacitance values is mainly focused on the synthesis of electrode materials with different structures and sizes. However, no studies have addressed using soluble inorganic salts directly as electrode materials. For the first time, we have reported that water-soluble CuCl2 electrodes show a fast and reversible redox reaction of Cu2+ ↔ Cu+ and deliver a very high specific pseudocapacitance, ~5442 F g−1. We have identified that the cation Cu2+ is responsible for achieving this big number. The chemical and crystallization transformation of the CuCl2 electrode is presented. Commercial inorganic salts can be used directly as electrodes neglecting complex synthesis procedures, which is an easily scalable and highly economical method. This method can be extended to a large variety of commercial inorganic salt electrodes following the guideline of ionic electronegativity.
Co-reporter:Kunfeng Chen and Dongfeng Xue
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 45) pp:19708-19714
Publication Date(Web):03 Oct 2013
DOI:10.1039/C3CP53787K
We reported a chemical reaction controlled mechanochemical route to synthesize mass CuO nanosheets by manual grinding in a mortar and pestle, which does not require any solvent, complex apparatus and techniques. The activation of chemical reactions by milling reactants was thus proved, and the energy from mechanical grinding promotes the fast formation of CuO nanoribbons. The resultant materials have preferential nanoscale ribbon-like morphology that can show large capacity and high cycle performance as lithium-ion battery anodes. After 50 cycles, the discharge capacity of CuO nanoribbon electrodes is 614.0 mA h g−1, with 93% retention of the reversible capacity. The thermodynamic reactions of the CuO battery showed size-dependent characterization. The microstructures of CuO nanosheets and reaction routes can be controlled by the ratio of NaOH/CuAc2 according to the chemical reactions involved. The intact nanoribbon structure, thin-layer, and hierarchical structures endow present CuO materials with high reversible capacity and excellent cycling performances. The simple, economical, and environmentally friendly mechanochemical route is of great interest in modern synthetic chemistry.
Co-reporter:Congting Sun and Dongfeng Xue
CrystEngComm 2013 vol. 15(Issue 48) pp:10445-10450
Publication Date(Web):02 Sep 2013
DOI:10.1039/C3CE41628C
In situ Attenuated Total Reflectance-Infrared (ATR-IR) spectroscopy was used to record and characterize the nucleation and crystal growth of KH2PO4 (KDP) from aqueous solution with pH = 1.01–6.96. In the crystallization of KDP, K+ and H2PO4− ions transform from hydrated ions to clusters and crystalline state. With increasing measurement time, the shift of v3(PO4) at 938 and 1145 cm−1 towards lower wavenumbers illustrated the breaking of hydrogen bonding between H2PO4− and H2O. IR bands of vO–H at 2370 cm−1 and βO–H at 1274 cm−1 indicated the formation of H2PO4− clusters possessing KDP lattice structural characteristics via hydrogen bonding. The disappearance of v3(PO4) at 1145 cm−1 indicated the chemical bonding between K+ and H2PO4− in the nucleation stage of KDP. In KDP supersaturated solution with pH = 2.02–6.96, H2PO4− groups with larger hydrated ionic radius initially form a framework structure, and then K+ ions with a relatively smaller hydrated ionic radius insert into the H2PO4− framework to form KDP. Moreover, the time needed for the appearance of KDP solids in IR measurement can be modified by varying the pH value of KDP supersaturated solution. Such an in situ visualization strategy can characterize the structural variations by spectroscopy, which deepens the insight into the nucleation and crystal growth from aqueous solution during the whole crystallization process.
Co-reporter:Congting Sun and Dongfeng Xue
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 34) pp:14414-14419
Publication Date(Web):01 Jul 2013
DOI:10.1039/C3CP51959G
We thermodynamically studied the size-dependent oxygen storage ability of nano-sized ceria by tracing the surface Ce/O ratio of octahedral particles with different diameters, from the viewpoint of lattice Ce and O in a CeO2 crystallographic structure. The high surface Ce/O ratio with small scale particle size has more excess surface Ce4+ ions, which allows ceria to have an increasing oxygen storage ability in a crystalline lattice. For the perfect octahedron growth shape of ceria, the nonstoichiometric surfaces can produce excess Ce4+ ions, Ce4+ ions can be stabilized by bonding with lattice oxygen, leading to an enhanced oxygen storage ability of ceria. With the increasing particle size, the surface Ce/O ratio approaches to 0.5 owing to the decreased contributions of atoms located at the edges and corners. When the octahedron diameter D = 0.55 nm, the surface Ce/O ratio can reach 0.75. When D = 7.58 nm, the surface Ce/O ratio decreases down to 0.51. If D ≥ 14.61 nm, the surface Ce/O ratios are equal to 0.5. The present study deepens the insight of the size-dependent oxygen storage ability of nano-sized ceria, focusing on the size-dependent excess Ce4+ on nonstoichiometric surfaces of ceria in thermodynamics.
Co-reporter:Keyan Li, Hui Xie, Jun Liu, Zengsheng Ma, Yichun Zhou and Dongfeng Xue
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 40) pp:17658-17663
Publication Date(Web):29 Aug 2013
DOI:10.1039/C3CP52997E
Toward engineering high performance anode alloys for Li-ion batteries, we proposed a useful method to quantitatively estimate the bulk modulus of binary alloys in terms of metallic electronegativity (EN), alloy composition and formula volume. On the basis of our proposed potential viewpoint, EN as a fundamental chemistry concept can be extended to be an important physical parameter to characterize the mechanical performance of Li–Si and Li–Sn alloys as anode materials for Li-ion batteries. The bulk modulus of binary alloys is linearly proportional to the combination of average metallic EN and atomic density of alloys. We calculated the bulk moduli of Li–Si and Li–Sn alloys with different Li concentrations, which can agree well with the reported data. The bulk modulus of Li–Si and Li–Sn alloys decreases with increasing Li concentration, leading to the elastic softening of the alloys, which is essentially caused by the decreased strength of constituent chemical bonds in alloys from the viewpoint of EN. This work provides a deep understanding of mechanical failure of Si and Sn anodes for Li-ion batteries, and permits the prediction of the composition dependent bulk modulus of various lithiated alloys on the basis of chemical formula, metallic EN and cell volume (or alloy density), with no structural details required.
Co-reporter:Congting Sun, Dongli Xu and Dongfeng Xue
CrystEngComm 2013 vol. 15(Issue 38) pp:7783-7791
Publication Date(Web):31 Jul 2013
DOI:10.1039/C3CE41249K
Crystallization of KDP-family crystals depends on the chemical bonding behavior of the crystal constituents in aqueous solution, which are sensitive to solution conditions. We applied in situ ATR-IR spectroscopy combined with a morphology-evolution calibration to declare the structural dynamics of NH4+ and H2PO4− during the NH4H2PO4 crystallization in aqueous solution with different concentrations and pH values. For unsaturated NH4H2PO4 solution, both the H2PO4− stretching vibration mode and NH4+ bending vibration mode are enhanced with increasing concentration. When the NH4H2PO4 solution becomes a saturated and then supersaturated and crystalline state, H2PO4− ions undergo hydrated dimerisation and polymerisation, which can be recorded by the appearance and red shift of the P–O⋯H–O–P in-plane bending vibration mode from 1250 to 1263 cm−1. During this process, hydrated NH4+ ions bind to the (H2PO4−)n frame, reflected by the splitting of the HN4+ bending vibration mode at 1450 and 1400 cm−1. For the supersaturated NH4H2PO4 solution, HPO42− and H2PO4− coexist in solution with increasing pH value up to 6.64, whereas H3PO4 and H2PO4− coexist with decreasing pH value down to 1.52. Such an in situ recording strategy is of particular value in studying system dynamics, and in general to monitor the solution concentrations and compositions before and during the crystallization process.
Co-reporter:Kunfeng Chen and Dongfeng Xue
The Journal of Physical Chemistry C 2013 Volume 117(Issue 44) pp:22576-22583
Publication Date(Web):October 8, 2013
DOI:10.1021/jp4081756
We demonstrated an efficient room-temperature chemical transformation route to CuO nanowires (NWs), from irregular particles to NWs coupled with a series of phase changes from CuCl, through Cu2(OH)3Cl, to Cu(OH)2, and finally to CuO. The room-temperature chemical transformation of Cu(OH)2 NW can reserve the initial NW morphology and made the synthesized CuO NW more active in electrochemical reactions. As the anode materials for lithium ion battery, these CuO NWs can exhibit a reversible capacity of 696.1 mAh g–1 after 40 cycles at the rate of 100 mA g–1. The high lithium-storage capacity can be ascribed to the unique structure of these CuO NWs with size of ∼10 nm and grain boundaries on the NWs surfaces, which show more active for the initial electrochemical reaction. CuO NWs and intermediate Cu(OH)2 NWs can also be fabricated as pseudocapacitor electrodes; in KOH electrolyte, their specific capacitances are 118 and 114 F g–1 at the current density of 1 A g–1. The present results indicate that the current room-temperature chemical transformation route is promising to produce advanced electrode materials for both lithium ion batteries and supercapacitors.
Co-reporter:Congting Sun and Dongfeng Xue
The Journal of Physical Chemistry C 2013 Volume 117(Issue 37) pp:19146-19153
Publication Date(Web):August 20, 2013
DOI:10.1021/jp407947s
In situ ATR-IR spectroscopy was used to identify the structural variations of NH4+ and H2PO4– during NH4H2PO4 (ADP) crystallization in aqueous solution with different pH values. For supersaturated ADP solution, the time needed for the appearance of ADP solids in IR measurement will be prolonged with increasing or decreasing solution pH value. When ADP crystallizes from the aqueous solution with pH = 0.98–5.00, H2PO4– groups with a larger hydrated ionic radius initially form a framework structure, and then NH4+ groups with a relatively smaller hydrated ionic radius insert into the H2PO4– framework. IR bands of the H-bond at 2387 cm–1 and a combination of PO4 vibration within the lattice at 1260 cm–1 indicated the formation of H2PO4– frameworks with ADP lattice structural characteristics via forming hydrogen bonding. The hydrogen bonding between NH4+ and H2PO4– can be indicated by the splitting of v4(NH4) at 1400 cm–1. IR spectra indicated that when the pH value increases up to 6.03, (NH4)2HPO4 instead of NH4H2PO4 was crystallized from the aqueous solution. Such an in situ recording strategy is of particular value in identifying the structural characteristics at both nucleation and crystal growth stages during the crystallization process.
Co-reporter:Xiaoyan Chen, Congting Sun, Sixin Wu and Dongfeng Xue
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 13) pp:NaN8842-8842
Publication Date(Web):2017/03/01
DOI:10.1039/C7CP00601B
Rare earth ions can be used to construct a variety of novel structures and are favorable to chemical bonding regulation and design. In this study, the chemical bonding paradigm between rare earth ions (Ln3+) and urea molecules in an aqueous solution can be tracked by the evolution of CO, NH2, and CN vibration bands during the urea nucleation stage. Rare earth ions such as La3+, Gd3+, and Lu3+ can manipulate the nucleation time of urea via regulating the nucleation-dependant N–CO⋯H–N hydrogen-bonding between urea molecules. Two types of chemical bondings between Ln3+ and urea molecules have been confirmed, which are Ln3+⋯OC–N and Ln3+⋯NH2−C. Compared with Ln3+⋯NH2−C, Ln3+ prefers to coordinate with the OC bond in urea. With a higher concentration of rare earth ions in the solution, some N–CO⋯H–N hydrogen bonds are broken as a consequence of the incorporation of Ln3+ into the lattice, resulting in the decreased symmetry of local urea molecules in the crystalline nuclei and the consequent Ln3+ concentration-dependent nucleation time of urea. Moreover, using the ionic electronegativity scale of Ln3+, the different effects of La3+, Gd3+, and Lu3+ on urea nucleation can be further distinguished. The present study provides basic data for unrevealing the chemical bonding regulation role of rare earth ions in the formation of hydrogen bonded materials, which may give insight into the design and fabrication of novel materials utilizing rare earth ions to adjust the chemical bonding process.
Co-reporter:Congting Sun and Dongfeng Xue
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 19) pp:NaN12413-12413
Publication Date(Web):2017/04/12
DOI:10.1039/C7CP01112A
Crystal growth is a dynamic physicochemical process, which depends on the multi-parameter synergetic control and directly determines the crystal features such as geometry and size. In this study, both thermodynamic and kinetic factors that determine inorganic single crystal growth are integrated by focusing on the mass transfer process at an interface. For the specific growth system, the integrated parameter is then classified to extract the critical control factors in anisotropic growth. The driving force of mass transfer essentially depends on the anisotropic chemical bonding architectures, leading to different concentration gradients along various [uvw] directions. Exquisitely controlling the chemical bonding architecture can therefore be used to regulate the mass transfer process of a compound in a straightforward manner, encompassing the origin of anisotropic growth as well as a variety of geometries in the formation of a multicomponent crystal.
Co-reporter:Kunfeng Chen, Shuyan Song and Dongfeng Xue
Journal of Materials Chemistry A 2015 - vol. 3(Issue 6) pp:NaN2453-2453
Publication Date(Web):2014/12/19
DOI:10.1039/C4TA06989G
Much progress about graphene has been made in the fields of physics, chemistry, material science, and electronics. Graphene's properties are mainly dependent on its geometric structures and synthesis methods. Various newly developed chemical methods have been designed to tailor graphene materials with specific functionalities, such as crystallization routes, which can be a new direction in graphene R&D. In this review, we focus on recent developments in the synthesis of graphene materials with specific structures and electrochemical performances by top-down routes. First, ice crystallization from water molecules within graphene oxide is discussed to form 3D graphene oxide aerogel and graphene aerogel with porous networks. Then we review an in situ electrochemical crystallization route to fabricate graphene/metal oxide aerogel electrode materials. The electrochemical properties of different structural graphene types are discussed as lithium-ion batteries and supercapacitors. Future challenges and current progress beyond graphene as an energy storage material have been highlighted.
Co-reporter:Kunfeng Chen and Dongfeng Xue
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 23) pp:NaN11172-11172
Publication Date(Web):2014/03/12
DOI:10.1039/C4CP00811A
An ex situ observation was made by XRD patterns, SEM and TEM images, as well as cyclic voltammogram curves of CuO/Cu integrated anodes for lithium ion batteries. For the first time, the existence of a Cu+ ion long-range transfer path was identified at the potential widow of 1.30–1.60 V during both charging and discharging processes. Both SEM and TEM images show that these nanowires networks hanging CuO nanoparticles provide a Cu+ diffusion path within our designed CuO/Cu integrated anode. This work provides new insights into the conversion reaction of inorganic anode materials, and can favor the development of high-performance conversion anodes for lithium-ion batteries.
Co-reporter:Congting Sun and Dongfeng Xue
Dalton Transactions 2017 - vol. 46(Issue 24) pp:NaN7896-7896
Publication Date(Web):2017/05/22
DOI:10.1039/C7DT01375B
REPO4 (RE = La, Gd, Lu, Y) serves as an excellent host lattice due to its stable physicochemical properties and optical inertia. Doping Gd3+(La3+) into LuPO4 can form mixed crystals, increasing the Gd3+(La3+) concentration will induce the phase transition from tetragonal to hexagonal lattices, and the variation of the local structure around the Ce3+ activator will influence its 5d-level position and consequently 5d → 4f radiation transition. This can be attributed to the synergy effect of rare earth ions in REPO4, however, the essential mechanism of such a synergy effect on the local structure and optical property is still poorly understood. Here, we study the synergy effect of rare earth ions on the phase transition and PL emission in a Ce3+:REPO4 system on the basis of the relationship between the composition-dependent local structure around Ce3+ and its PL emission properties from a molecular view. The competition between Lu3+ and Gd3+(La3+) in REPO4 not only influences the relative atomic position but also varies the symmetry of anion groups. Infrared absorption bands indicate that the activation of P−O bonding promotes phase transition and enhances PL emission intensity. The PL emission intensity of Ce3+ is higher in a REPO4 host with a lower site symmetry PO43− group (C2) than that with a higher site symmetry PO43− group (D2d). An increased disorder degree in Ce:GdxLu1−xPO4 mixed crystals leads to the shift of the 5d-level of Ce3+ towards a higher position, resulting in the blue shift of the PL emission wavelength. Moreover, the 5d → 4f emission of Ce3+ may also be modulated towards a larger wavelength via substituting the cation site with larger-radius cations under a particular crystallographic structure in REPO4. Our results highlight the importance of disordered local structures as well as activated anion groups in the enhanced PL emission of Ce3+ activators in a host lattice.
Co-reporter:Kunfeng Chen, Shuyan Song, Fei Liu and Dongfeng Xue
Chemical Society Reviews 2015 - vol. 44(Issue 17) pp:NaN6257-6257
Publication Date(Web):2015/06/08
DOI:10.1039/C5CS00147A
There are many practical challenges in the use of graphene materials as active components in electrochemical energy storage devices. Graphene has a much lower capacitance than the theoretical capacitance of 550 F g−1 for supercapacitors and 744 mA h g−1 for lithium ion batteries. The macroporous nature of graphene limits its volumetric energy density and the low packing density of graphene-based electrodes prevents its use in commercial applications. Increases in the capacity, energy density and power density of electroactive graphene materials are strongly dependent on their microstructural properties, such as the number of defects, stacking, the use of composite materials, conductivity, the specific surface area and the packing density. The structural design of graphene electrode materials is achieved via six main strategies: the design of non-stacking and three-dimensional graphene; the synthesis of highly packed graphene; the production of graphene with a high specific surface area and high conductivity; the control of defects; functionalization with O, N, B or P heteroatoms; and the formation of graphene composites. These methodologies of structural design are needed for fast electrical charge storage/transfer and the transport of electrolyte ions (Li+, H+, K+, Na+) in graphene electrodes. We critically review state-of-the-art progress in the optimization of the electrochemical performance of graphene-based electrode materials. The structure of graphene needs to be designed to develop novel electrochemical energy storage devices that approach the theoretical charge limit of graphene and to deliver electrical energy rapidly and efficiently.
Co-reporter:Keyan Li, Hui Xie, Jun Liu, Zengsheng Ma, Yichun Zhou and Dongfeng Xue
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 40) pp:NaN17663-17663
Publication Date(Web):2013/08/29
DOI:10.1039/C3CP52997E
Toward engineering high performance anode alloys for Li-ion batteries, we proposed a useful method to quantitatively estimate the bulk modulus of binary alloys in terms of metallic electronegativity (EN), alloy composition and formula volume. On the basis of our proposed potential viewpoint, EN as a fundamental chemistry concept can be extended to be an important physical parameter to characterize the mechanical performance of Li–Si and Li–Sn alloys as anode materials for Li-ion batteries. The bulk modulus of binary alloys is linearly proportional to the combination of average metallic EN and atomic density of alloys. We calculated the bulk moduli of Li–Si and Li–Sn alloys with different Li concentrations, which can agree well with the reported data. The bulk modulus of Li–Si and Li–Sn alloys decreases with increasing Li concentration, leading to the elastic softening of the alloys, which is essentially caused by the decreased strength of constituent chemical bonds in alloys from the viewpoint of EN. This work provides a deep understanding of mechanical failure of Si and Sn anodes for Li-ion batteries, and permits the prediction of the composition dependent bulk modulus of various lithiated alloys on the basis of chemical formula, metallic EN and cell volume (or alloy density), with no structural details required.
Co-reporter:Kunfeng Chen and Dongfeng Xue
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 45) pp:NaN19714-19714
Publication Date(Web):2013/10/03
DOI:10.1039/C3CP53787K
We reported a chemical reaction controlled mechanochemical route to synthesize mass CuO nanosheets by manual grinding in a mortar and pestle, which does not require any solvent, complex apparatus and techniques. The activation of chemical reactions by milling reactants was thus proved, and the energy from mechanical grinding promotes the fast formation of CuO nanoribbons. The resultant materials have preferential nanoscale ribbon-like morphology that can show large capacity and high cycle performance as lithium-ion battery anodes. After 50 cycles, the discharge capacity of CuO nanoribbon electrodes is 614.0 mA h g−1, with 93% retention of the reversible capacity. The thermodynamic reactions of the CuO battery showed size-dependent characterization. The microstructures of CuO nanosheets and reaction routes can be controlled by the ratio of NaOH/CuAc2 according to the chemical reactions involved. The intact nanoribbon structure, thin-layer, and hierarchical structures endow present CuO materials with high reversible capacity and excellent cycling performances. The simple, economical, and environmentally friendly mechanochemical route is of great interest in modern synthetic chemistry.
Co-reporter:Congting Sun and Dongfeng Xue
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 34) pp:NaN14419-14419
Publication Date(Web):2013/07/01
DOI:10.1039/C3CP51959G
We thermodynamically studied the size-dependent oxygen storage ability of nano-sized ceria by tracing the surface Ce/O ratio of octahedral particles with different diameters, from the viewpoint of lattice Ce and O in a CeO2 crystallographic structure. The high surface Ce/O ratio with small scale particle size has more excess surface Ce4+ ions, which allows ceria to have an increasing oxygen storage ability in a crystalline lattice. For the perfect octahedron growth shape of ceria, the nonstoichiometric surfaces can produce excess Ce4+ ions, Ce4+ ions can be stabilized by bonding with lattice oxygen, leading to an enhanced oxygen storage ability of ceria. With the increasing particle size, the surface Ce/O ratio approaches to 0.5 owing to the decreased contributions of atoms located at the edges and corners. When the octahedron diameter D = 0.55 nm, the surface Ce/O ratio can reach 0.75. When D = 7.58 nm, the surface Ce/O ratio decreases down to 0.51. If D ≥ 14.61 nm, the surface Ce/O ratios are equal to 0.5. The present study deepens the insight of the size-dependent oxygen storage ability of nano-sized ceria, focusing on the size-dependent excess Ce4+ on nonstoichiometric surfaces of ceria in thermodynamics.
Co-reporter:Kunfeng Chen, Congting Sun and Dongfeng Xue
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 2) pp:NaN750-750
Publication Date(Web):2014/11/10
DOI:10.1039/C4CP03888F
Advances in materials have preceded almost every major technological leap since the beginning of civilization. On the nanoscale and microscale, mastery over the morphology, size, and structure of a material enables control of its properties and enhancement of its usefulness for a given application, such as energy storage. In this review paper, our aim is to present a review of morphology engineering of high performance oxide electrode materials for electrochemical energy storage. We begin with the chemical bonding theory of single crystal growth to direct the growth of morphology-controllable materials. We then focus on the growth of various morphologies of binary oxides and their electrochemical performances for lithium ion batteries and supercapacitors. The morphology–performance relationships are elaborated by selecting examples in which there is already reasonable understanding for this relationship. Based on these comprehensive analyses, we proposed colloidal supercapacitor systems beyond morphology control on the basis of system- and ion-level design. We conclude this article with personal perspectives on the directions toward which future research in this field might take.
Co-reporter:Kunfeng Chen and Dongfeng Xue
Journal of Materials Chemistry A 2016 - vol. 4(Issue 20) pp:NaN7537-7537
Publication Date(Web):2016/04/11
DOI:10.1039/C6TA01527A
Materials chemistry focuses on all aspects of the production of electrode materials or the properties or applications of materials related to energy storage, which thus plays an important role in the field of energy storage. Electrochemical energy storage includes the conversion reaction between chemical energy and electric energy, with the electric energy being stored in chemical bonds of electrode materials of both battery and pseudocapacitor types. Energy density, power density and safety of these devices, i.e. lithium ion batteries and supercapacitors, are mostly dependent on the electrode materials with high electroactivity, high electron/ion conductivity, and high structural/electrochemical stability. Following the function-directed materials design rule, we can select appropriate elements, chemical bonds, crystal structures, and morphologies of those materials toward high electrochemical performances. In this review, we summarize, from both theoretical and experimental viewpoints of materials chemistry, recent advances in designing electrode materials from element and structure selections to final morphology selection. Electronegativity, atom radius, chemical bonding behavior, and oxidation state have been identified as controllable materials properties to synthesize high-performance electrode materials. This review provides general materials chemistry rules to rationally design electrode materials with improved electrochemical performance.