Co-reporter:Jamie A. Leitch, Yunas Bhonoah, and Christopher G. Frost
ACS Catalysis September 1, 2017 Volume 7(Issue 9) pp:5618-5618
Publication Date(Web):July 11, 2017
DOI:10.1021/acscatal.7b01785
The indole scaffold will continue to play a vital part in the future of drug discovery and agrochemical development. Because of this, the necessity for elegant techniques to enable selective C–H functionalization is vast. Early developments have led to primarily C2 and C3 functionalization because of the inherent reactivity of the pyrrole ring. Despite this, elegant methods have been developed to enable selective C–H functionalization on the benzenoid moiety at C4, C5, C6, and C7. This review focuses on the contributions made in benzenoid C–H functionalization of indoles and other related heteroaromatics such as carbazoles.Keywords: C−H functionalization; heteroaromatics; homogeneous catalysis; indole; regioselectivity;
Co-reporter:Jamie A. Leitch, Claire L. McMullin, Mary F. Mahon, Yunas Bhonoah, and Christopher G. Frost
ACS Catalysis April 7, 2017 Volume 7(Issue 4) pp:2616-2616
Publication Date(Web):March 6, 2017
DOI:10.1021/acscatal.7b00038
The site-selective functionalization of an indole template offers exciting possibilities for the derivatization of molecules with useful biological properties. Herein, we report the remote C6-selective C–H alkylation of indole derivatives enabled by dual cyclometalation/redox ruthenium catalysis. Remote alkylation was achieved using N-pyrimidinyl indoles with an ancillary ester directing group at the C3 position. This ancillary directing group proved pivotal to reactivity at C6, with yields up to 92% achieved. A one-pot procedure to install this directing group followed by remote C6 functionalization has also been reported; both processes are shown to proceed via ruthenium redox catalysis. Computationally calculated Fukui indices elucidated that the C6 position was the most reactive vacant C–H site toward potential functionalization. When this investigation was coupled with deuterium incorporation studies, a C2-cyclometalation/remote σ-activation pathway was deduced.Keywords: heteroaromatics; homogeneous catalysis; indole; remote functionalization; ruthenium;
Co-reporter:William Mahy;Sinéad Cabezas-Hayes;Gabriele Kociok-Köhn
European Journal of Organic Chemistry 2017 Volume 2017(Issue 43) pp:6441-6444
Publication Date(Web):2017/11/24
DOI:10.1002/ejoc.201701181
This work describes an operationally simple catalytic synthesis of cyclic S-thiocarbonates with predictable regioselectivity in good yields. The reaction utilizes substrates derived from ubiquitous 1,2-diols in an atom economical intramolecular rearrangement, catalysed by an inexpensive and simple catalyst–ligand system. A crystal structure is presented that clearly confirms the regioselectivity of the reaction.
Co-reporter:Andrew J. Paterson;Callum J. Heron;Claire L. McMullin;Mary F. Mahon;Neil J. Press
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 28) pp:5993-6000
Publication Date(Web):2017/07/19
DOI:10.1039/C7OB01192J
A catalytic meta selective C–H alkylation of arenes is described using a wide range of α-halo carbonyls as coupling partners. Previously unreported primary alkylations with high meta selectivity have been enabled by this methodology whereas using straight chain alkyl halides affords ortho substituted products. Mechanistic analysis reveals an activation pathway whereby cyclometalation with a ruthenium(II) complex activates the substrate molecule and is responsible for the meta selectivity observed. A distinct second activation of the coupling partner allows site selective reaction between both components.
Co-reporter:Jamie A. Leitch
Chemical Society Reviews 2017 vol. 46(Issue 23) pp:7145-7153
Publication Date(Web):2017/11/27
DOI:10.1039/C7CS00496F
The search for selective C–H functionalisation has enabled some of the most elegant techniques in modern catalysis. Herein, we review the rapidly expanding field of ruthenium catalysed σ-activation as a tool in the selective meta-C–H functionalisation of arenes.
Co-reporter:Jamie A. Leitch;Callum J. Heron;Janette McKnight;Gabriele Kociok-Köhn;Yunas Bhonoah
Chemical Communications 2017 vol. 53(Issue 97) pp:13039-13042
Publication Date(Web):2017/12/05
DOI:10.1039/C7CC07606A
We report the C4-selective C–H alkylation of carbazole derivatives furnished with a pyrimidine directing group at N9. This was realized using ruthenium catalyzed σ-activation methodology, whereby C–H activation at C1 enables the interaction of this ruthenacycle, at the para position to the metal center, with tertiary alkyl radicals.
Co-reporter:Jamie A. Leitch;Dr. Claire L. McMullin;Dr. Andrew J. Paterson;Dr. Mary F. Mahon;Dr. Yunas Bhonoah; Christopher G. Frost
Angewandte Chemie 2017 Volume 129(Issue 47) pp:15327-15331
Publication Date(Web):2017/11/20
DOI:10.1002/ange.201708961
AbstractThe para-selective C−H alkylation of aniline derivatives furnished with a pyrimidine auxiliary is herein reported. This reaction is proposed to take place via an N−H-activated cyclometalate formed in situ. Experimental and DFT mechanistic studies elucidate a dual role of the ruthenium catalyst. Here the ruthenium catalyst can undergo cyclometalation by N−H metalation (as opposed to C−H metalation in meta-selective processes) and form a redox active ruthenium species, to enable site-selective radical addition at the para position.
Co-reporter:Jamie A. Leitch;Dr. Claire L. McMullin;Dr. Andrew J. Paterson;Dr. Mary F. Mahon;Dr. Yunas Bhonoah; Christopher G. Frost
Angewandte Chemie International Edition 2017 Volume 56(Issue 47) pp:15131-15135
Publication Date(Web):2017/11/20
DOI:10.1002/anie.201708961
AbstractThe para-selective C−H alkylation of aniline derivatives furnished with a pyrimidine auxiliary is herein reported. This reaction is proposed to take place via an N−H-activated cyclometalate formed in situ. Experimental and DFT mechanistic studies elucidate a dual role of the ruthenium catalyst. Here the ruthenium catalyst can undergo cyclometalation by N−H metalation (as opposed to C−H metalation in meta-selective processes) and form a redox active ruthenium species, to enable site-selective radical addition at the para position.
Co-reporter:Jamie A. Leitch, Philippe B. Wilson, Claire L. McMullin, Mary F. Mahon, Yunas Bhonoah, Ian H. Williams, and Christopher G. Frost
ACS Catalysis 2016 Volume 6(Issue 8) pp:5520
Publication Date(Web):July 14, 2016
DOI:10.1021/acscatal.6b01370
Herein, we report the ruthenium-catalyzed ortho C–H alkenylation of a wide range of N-aryloxazolidinone scaffolds. Alkenylation was achieved with complete monoselectivity with a scope of 27 examples in 2-MeTHF. Yields ranged from 23 to 94%, producing highly decorated oxazolidinone scaffolds. A kinetically relevant C–H cleavage was also observed with a kinetic isotope effect (KIE) of ∼2. Density functional theory calculations provided information about mechanism, detailing the β-hydride elimination as the most energetically challenging step of 13.5 kcal mol–1. In-depth computational kinetic studies also predicted a KIE of 2.17 for C–H cleavage and an intrinsic KIE for the reaction of 2.22, in line with the experimentally observed value.Keywords: C−H activation; DFT; heterocycles; homogeneous catalysis; kinetic isotope effect; ruthenium
Co-reporter:William Mahy;Jamie A. Leitch
European Journal of Organic Chemistry 2016 Volume 2016( Issue 7) pp:1305-1313
Publication Date(Web):
DOI:10.1002/ejoc.201600033
Abstract
The total synthesis of oxazolidinone-based pharmaceuticals, linezolid, tedizolid and rivaroxaban is reported. They are synthesized using a recently reported copper-catalyzed one-pot cyclization and arylation as the key step to construct the N-aryloxazolidinone core. Active pharmaceutical ingredients (API) were synthesized from a common synthetic pool of a simple protected amino alcohol in 22 %, 61 % and 40 % total synthesis yields, respectively.
Co-reporter:Jamie A. Leitch, Hans P. Cook, Yunas Bhonoah, and Christopher G. Frost
The Journal of Organic Chemistry 2016 Volume 81(Issue 20) pp:10081-10087
Publication Date(Web):September 28, 2016
DOI:10.1021/acs.joc.6b02073
Ruthenium(II)-catalyzed C–H functionalization of N-arylhydantoins is herein described. The biologically relevant hydantoin (imidazolidinedione) heterocycle functions as a weakly coordinating directing group in a C–H alkenylation reaction. The reaction gave a wide scope of 23 examples with yields up to 94% in the green solvent 2-MeTHF. Functionalization of API nilutamide (antiandrogen) is also reported. The use of the succinimide heterocycle as a directing group is also demonstrated in modest yields.
Co-reporter:Sean Goggins, Barrie J. Marsh, Anneke T. Lubben and Christopher G. Frost
Chemical Science 2015 vol. 6(Issue 8) pp:4978-4985
Publication Date(Web):15 Jun 2015
DOI:10.1039/C5SC01588J
Signal transduction and signal amplification are both important mechanisms used within biological signalling pathways. Inspired by this process, we have developed a signal amplification methodology that utilises the selectivity and high activity of enzymes in combination with the robustness and generality of an organometallic catalyst, achieving a hybrid biological and synthetic catalyst cascade. A proligand enzyme substrate was designed to selectively self-immolate in the presence of the enzyme to release a ligand that can bind to a metal pre-catalyst and accelerate the rate of a transfer hydrogenation reaction. Enzyme-triggered catalytic signal amplification was then applied to a range of catalyst substrates demonstrating that signal amplification and signal transduction can both be achieved through this methodology.
Co-reporter:Andrew J. Paterson, Sahra St John-Campbell, Mary F. Mahon, Neil J. Press and Christopher G. Frost
Chemical Communications 2015 vol. 51(Issue 64) pp:12807-12810
Publication Date(Web):06 Jul 2015
DOI:10.1039/C5CC03951G
A catalytic meta-selective C–H functionalization of 2-phenylpyridines using a range of tertiary halides is described. The protocol is simple to perform and uses commercially available reagents to construct challenging quaternary carbon centres in a regioselective manner. Preliminary studies suggest the C–H functionalization proceeds through a radical process directed via a remote σ-activation.
Co-reporter:Sean Goggins, Christophe Naz, Barrie J. Marsh and Christopher G. Frost
Chemical Communications 2015 vol. 51(Issue 3) pp:561-564
Publication Date(Web):10 Nov 2014
DOI:10.1039/C4CC07693A
A novel ferrocene-derived substrate for the ratiometric electrochemical detection of alkaline phosphatase (ALP) was designed and synthesised. It was demonstrated to be an excellent electrochemical substrate for the ALP-labelled enzyme-linked immunosorbent assay (ELISA).
Co-reporter:Zhugen Yang, Barbara Kasprzyk-Hordern, Christopher G. Frost, Pedro Estrela, and Kevin V. Thomas
Environmental Science & Technology 2015 Volume 49(Issue 10) pp:5845
Publication Date(Web):May 8, 2015
DOI:10.1021/acs.est.5b01434
Co-reporter:Zhugen Yang, Marc Anglès d’Auriac, Sean Goggins, Barbara Kasprzyk-Hordern, Kevin V. Thomas, Christopher G. Frost, and Pedro Estrela
Environmental Science & Technology 2015 Volume 49(Issue 9) pp:5609
Publication Date(Web):April 8, 2015
DOI:10.1021/acs.est.5b00637
A new label-free electrochemical DNA (E-DNA) biosensor using a custom synthesized ferrocenyl (Fc) double-stranded DNA intercalator as a redox marker is presented. Single-stranded DNA (ssDNA) was co-immobilized on gold electrodes with 6-mecarpto-hexanol to control the surface density of the ssDNA probe, and hybridized with complementary DNA. The binding of the Fc intercalator to dsDNA was measured by differential pulse voltammetry. This new biosensor was optimized to allow the detection of single base pair mismatched sequences, able to detect as low as 10 pM target ssDNA with a dynamic range from 10 pM to 100 nM. DNA extracted from wastewater was analyzed by quantitative polymerase chain reaction targeting human-specific mitochondrial DNA (mtDNA). The aim of this approach is to enable the analysis of population biomarkers in wastewater for the evaluation of public health using wastewater-based epidemiology (WBE). The E-DNA biosensor was employed to detect human-specific mtDNA from wastewater before and after PCR amplification. The results demonstrate the feasibility of detecting human DNA biomarkers in wastewater using the developed biosensor, which may allow the further development of DNA population biomarkers for public health using WBE.
Co-reporter:Zhugen Yang, Barbara Kasprzyk-Hordern, Sean Goggins, Christopher G. Frost and Pedro Estrela
Analyst 2015 vol. 140(Issue 8) pp:2628-2633
Publication Date(Web):24 Feb 2015
DOI:10.1039/C4AN02277G
We report on a novel strategy for DNA aptamer immobilization to develop sensitive electrochemical detection of a protein biomarker, with prostate specific antigen (PSA) as a case biomarker. Thiolated single-stranded DNA (ssDNA) was co-immobilized with 3-mercapto-1-propanol on gold electrodes, and used as a scaffold for DNA aptamer attachment through hybridization of the aptamer overhang (so-called “DNA-directed immobilization aptamer sensors”, DDIAS). In the approach, the complementary DNA aptamer against PSA was assembled by the probe ssDNA onto the electrode to detect PSA; or the probe ssDNA directly hybridized with a complementary DNA aptamer/PSA complex following their pre-incubation in solution, so-called ‘on-chip’ and ‘in-solution’ methods, respectively. A double stranded DNA intercalator with a ferrocenyl (Fc) redox marker was synthesized to evaluate the feasibility of the strategy. The results demonstrate that the ‘in-solution’ method offers a favourable medium (in a homogeneous solution) for the binding between the aptamer and PSA, which shows to be more efficient than the ‘on-chip’ approach. DDIAS shows promising analytical performance under optimized conditions, with a limit of detection in the range of fM and low non-specific adsorption.
Co-reporter:William Mahy;Dr. Pawel Plucinski;Dr. Jesús Jover;Dr. Christopher G. Frost
Angewandte Chemie International Edition 2015 Volume 54( Issue 37) pp:10944-10948
Publication Date(Web):
DOI:10.1002/anie.201505280
Abstract
A practical ruthenium-catalyzed O- to S-alkyl migration affords structurally diverse thiooxazolidinones in excellent yields. Our studies suggest this catalytic transformation proceeds through a pseudoreversible radical pathway drawing mechanistic parallels to the classic Barton–McCombie reaction.
Co-reporter:William Mahy;Dr. Pawel Plucinski;Dr. Jesús Jover;Dr. Christopher G. Frost
Angewandte Chemie 2015 Volume 127( Issue 37) pp:11094-11098
Publication Date(Web):
DOI:10.1002/ange.201505280
Abstract
A practical ruthenium-catalyzed O- to S-alkyl migration affords structurally diverse thiooxazolidinones in excellent yields. Our studies suggest this catalytic transformation proceeds through a pseudoreversible radical pathway drawing mechanistic parallels to the classic Barton–McCombie reaction.
Co-reporter:William Mahy, Pawel K. Plucinski, and Christopher G. Frost
Organic Letters 2014 Volume 16(Issue 19) pp:5020-5023
Publication Date(Web):September 16, 2014
DOI:10.1021/ol502322c
An efficient sequential intramolecular cyclization of amino alcohol carbamates followed by Cu-catalyzed cross-coupling with aryl iodides under mild conditions has been developed. The reaction occurred in good yields and tolerated aryl iodides containing functionalities such as nitriles, ketones, ethers, and halogens. Heteroaryl iodides and substituted amino alcohol carbamates were also well tolerated.
Co-reporter:William R. Reynolds, Pawel Plucinski and Christopher G. Frost
Catalysis Science & Technology 2014 vol. 4(Issue 4) pp:948-954
Publication Date(Web):23 Jan 2014
DOI:10.1039/C3CY00836C
Two novel polymer encapsulated silica supported palladium catalysts have been prepared and shown to be highly active, robust and recyclable catalysts for Heck and Suzuki–Miyaura cross coupling reactions under continuous flow conditions. They were demonstrated to give excellent conversions of both electron rich and electron deficient substrates and the catalysts were used for over 50 hours continuous operation without any appreciable decrease in catalytic activity and low levels of Pd leaching measured by ICP-MS.
Co-reporter:Sean Goggins, Eleanor Rosevere, Clément Bellini, Joseph C. Allen, Barrie J. Marsh, Mary F. Mahon and Christopher G. Frost
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 1) pp:47-52
Publication Date(Web):07 Nov 2013
DOI:10.1039/C3OB42099J
The synthesis of a range of novel silyl-protected dioxaborinanes as a column- and bench-stable boron reagent were found to be advantageous to achieving good yields in palladium-catalysed cross-coupling reactions under standard conditions.
Co-reporter:Barrie J. Marsh, Lauren Hampton, Sean Goggins and Christopher G. Frost
New Journal of Chemistry 2014 vol. 38(Issue 11) pp:5260-5263
Publication Date(Web):07 Aug 2014
DOI:10.1039/C4NJ01050G
Electron-withdrawing or -donating groups are known to directly affect the Fe(III/II) formal potential of ferrocene derivatives by affecting their energy levels relative to the vacuum level. However, perhaps surprisingly, also more subtle indirect “tuning” of the formal potential is possible by changing the “dielectric environment”. This is demonstrated here by systematically changing the chain length of alkyl-chain derivatives. The resulting formal potentials are shown to be correlated to the hydrophobicity of the ferrocene molecule which can be used to predict the redox potential of a ferrocene.
Co-reporter:Po Man Liu and Christopher G. Frost
Organic Letters 2013 Volume 15(Issue 22) pp:5862-5865
Publication Date(Web):November 6, 2013
DOI:10.1021/ol402936c
A ruthenium-catalyzed C–H acylation of arylpyrazoles with a variety of acyl chlorides is described. The acylation reaction exhibits good regioselectivity and both aromatic and aliphatic acyl chlorides can be effectively coupled to the arylpyrazoles at the ortho-position.
Co-reporter:Paula M. T. Ferreira;Luís S. Monteiro;Goreti Pereira;Elisabete M. S. Castanheira
European Journal of Organic Chemistry 2013 Volume 2013( Issue 3) pp:550-556
Publication Date(Web):
DOI:10.1002/ejoc.201201198
Abstract
Several β-arylalanine derivatives containing fluorescent groups were prepared in good yields using a rhodium-catalysed conjugate addition of arylboronic acids to N,N-diprotected and N-monoprotected dehydroalanines. The best conditions for these reactions required the use of an excess of arylboronic acid (4 equiv.), [Rh(COD)2]BF4 as catalyst, and CsF as base in dioxane/H2O (10:1) at 110 °C. These conditions were also applied to several dipeptides with dehydroalanine residues. The photophysical properties of some of the β-arylalanines were studied in three solvents with different polarities. Due to the absence of the α,β-double bond, the absorption and fluorescence emission of the new compounds are dominated by the photophysical properties of the polycyclic aromatic fluorophores (naphthalene, phenanthrene, and pyrene). Considering the relatively high fluorescence quantum yield of these compounds, some of them may be useful as fluorescent markers for peptides and proteins.
Co-reporter:Joseph C. Allen, Gabriele Kociok-Köhn and Christopher G. Frost
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 1) pp:32-35
Publication Date(Web):05 Oct 2011
DOI:10.1039/C1OB06586F
The rhodium-catalysed enantioselective 1,4-addition of organoboron reagents to arylidene Meldrum's acids as acceptors, allows convenient access to 4-arylchroman-2-ones with good to excellent levels of enantioselectivity. The use of silyl-protected dioxaborinanes as donors was found to be advantageous to achieving good yields of product under anhydrous conditions.
Co-reporter:Ourida Saidi ; Jameel Marafie ; Araminta E. W. Ledger ; Po Man Liu ; Mary F. Mahon ; Gabriele Kociok-Köhn ; Michael K. Whittlesey
Journal of the American Chemical Society 2011 Volume 133(Issue 48) pp:19298-19301
Publication Date(Web):November 2, 2011
DOI:10.1021/ja208286b
A selective catalytic meta sulfonation of 2-phenylpyridines was found to occur in the presence of (arene)ruthenium(II) complexes upon reaction with sulfonyl chlorides. The 2-pyridyl group facilitates the formation of a stable Ru–Caryl σ bond that induces a strong para-directing effect. Electrophilic aromatic substitution proceeds with the sulfonyl chloride to furnish a sulfone at the position meta to the chelating group. This new catalytic process offers access to atypical regioselectivity for reactions involving chelation-assisted cyclometalation.
Co-reporter:Hannah J. Edwards, Jonathan D. Hargrave, Stephen D. Penrose and Christopher G. Frost
Chemical Society Reviews 2010 vol. 39(Issue 6) pp:2093-2105
Publication Date(Web):21 Apr 2010
DOI:10.1039/B919762C
The rhodium catalysed conjugate addition of organometallics to activated alkenes is a powerful synthetic tool for establishing new carbon–carbon bonds often with high stereoselectivity. The introduction of a practical, efficient method for introducing functionalised aryl and alkenyl fragments with predictable stereocontrol has caught the attention of synthetic chemists and emerging examples are growing in number and complexity. In this tutorial review, we document notable advances in the application of rhodium catalysed conjugate addition processes within the context of synthesis of complex molecules and intermediates in drug discovery. The chosen examples illustrate important issues regarding scope, selectivity and reactivity that will help guide the selection of appropriate donor and acceptor to achieve the desired carbon–carbon bond construction when planning new synthetic routes.
Co-reporter:Christopher G. Frost and Lynsey Mutton
Green Chemistry 2010 vol. 12(Issue 10) pp:1687-1703
Publication Date(Web):17 Sep 2010
DOI:10.1039/C0GC00133C
The application of heterogeneous catalysis in conjunction with microreactor technology can facilitate a cleaner and scalable flow methodology for organic synthesis. In this tutorial review we present recent advances in the design of supported catalysts for emerging synthetic applications within microreactor technology. Specifically, transition metal catalysts such as palladium, copper, ruthenium, and nickel are described on silica, monolithic, magnetic nanoparticles and polymer supports. These catalysts have been utilised to promote a range of reactions including Heck, Sonogashira, Suzuki, Kumada, olefin metathesis, hydrogenation and benzannulation reactions.
Co-reporter:Jonathan D. Hargrave, Gerwyn Bish, Gabriele Kociok Köhn and Christopher G. Frost
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 22) pp:5120-5125
Publication Date(Web):08 Sep 2010
DOI:10.1039/C0OB00158A
The rhodium-catalysed conjugate addition of arylboronic acids to an enantiopure acceptor derived from (R)-S-methylcysteine proceeds under substrate control to provide a range of functionalised phenylalanine derivatives with excellent stereocontrol via a highly diastereoselective protonation.
Co-reporter:James R. White, Gareth J. Price, Stefanie Schiffers, Paul R. Raithby, Pawel K. Plucinski, Christopher G. Frost
Tetrahedron Letters 2010 Volume 51(Issue 30) pp:3913-3917
Publication Date(Web):28 July 2010
DOI:10.1016/j.tetlet.2010.05.104
The stable benzylazido-boronate ester 1 is presented as an example of a dual-functional linker that allows the synthetically valuable boronate motif to be clicked onto other molecules under mild conditions. The utility of the azido-boronate motif as a modular building block is demonstrated in the rapid synthesis of drug-like structures employing sequential catalytic azide–alkyne cycloaddition and Suzuki coupling reactions.The utility of a bench-stable azido-boronate motif as a useful modular building block is demonstrated in the rapid synthesis of drug-like structures employing sequential catalytic azide–alkyne cycloaddition and Suzuki coupling reactions.
Co-reporter:JonathanD. Hargrave Dr.;JosephC. Allen;Gabriele Kociok-Köhn Dr.;Gerwyn Bish;ChristopherG. Frost Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 10) pp:1825-1829
Publication Date(Web):
DOI:10.1002/anie.200907067
Co-reporter:JonathanD. Hargrave Dr.;JosephC. Allen;Gabriele Kociok-Köhn Dr.;Gerwyn Bish;ChristopherG. Frost Dr.
Angewandte Chemie 2010 Volume 122( Issue 10) pp:1869-1873
Publication Date(Web):
DOI:10.1002/ange.200907067
Co-reporter:JonathanD. Hargrave Dr.;JosephC. Allen ;ChristopherG. Frost Dr.
Chemistry – An Asian Journal 2010 Volume 5( Issue 3) pp:386-396
Publication Date(Web):
DOI:10.1002/asia.200900512
Abstract
The rhodium-catalyzed conjugate addition of organoboron reagents to alkene acceptors has emerged as an important methodology in organic synthesis. The process results in the formation of new carbon–carbon bonds, and in principle, can create new stereocenters at both the α and β carbon atoms of the acceptor. Other organometallics are known to participate in the key transmetalation to rhodium and are becoming more established in stereoselective synthesis (notably arylzinc reagents). This Focus Review will describe recent developments in the use of alternative organometallic donors to organoboron reagents in rhodium-catalyzed conjugate addition reactions and their growing applications in synthesis.
Co-reporter:Ludivine Zoute, Gabriele Kociok-Köhn and Christopher G. Frost
Organic Letters 2009 Volume 11(Issue 12) pp:2491-2494
Publication Date(Web):May 22, 2009
DOI:10.1021/ol900843a
The rhodium-catalyzed 1,4-addition of arylboronic acids to an enantiopure heterocyclic acceptor proceeds under ligand control to effect an asymmetric synthesis of functionalized pyrrolizidinones. The protocol allows convenient access to all four stereoisomers of pyrrolizidinone 3a (Ar = Ph) by appropriate selection of substrate and catalyst.
Co-reporter:James R. White, Gareth J. Price, Pawel K. Plucinski, Christopher G. Frost
Tetrahedron Letters 2009 50(52) pp: 7365-7368
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.10.082
Co-reporter:Jérôme Le Nôtre, Joseph C. Allen and Christopher G. Frost
Chemical Communications 2008 (Issue 32) pp:3795-3797
Publication Date(Web):23 Jun 2008
DOI:10.1039/B806098C
The enantioselective 1,4-addition of 2-(substituted)thienylzinc and 2-furanylzinc reagents has been achieved (up to 99 : 1 er) using a complex derived from [Rh(C2H4)2Cl]2 and Me-DUPHOS.
Co-reporter:Christopher G. Frost, Stephen D. Penrose and Robert Gleave
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 23) pp:4340-4347
Publication Date(Web):18 Oct 2008
DOI:10.1039/B812897A
The concise enantioselective synthesis of hermitamides A and B is presented utilising a rhodium catalysed conjugate addition reaction to introduce the side chain and chiral information in a single step via an alkenyltrifluoroborate salt.
Co-reporter:Christopher J. Chapman, Ai Matsuno, Christopher G. Frost and Michael C. Willis
Chemical Communications 2007 (Issue 38) pp:3903-3905
Publication Date(Web):15 Aug 2007
DOI:10.1039/B711533D
The site-selective modification of peptides containing dehydroalanine, tyrosine and tryptophan residues has been achieved using rhodium catalysed conjugate additions or palladium catalysed aryl-amination and -etherification reactions.
Co-reporter:Jérôme Le Nôtre;David van Mele;Christopher G. Frost
Advanced Synthesis & Catalysis 2007 Volume 349(Issue 3) pp:
Publication Date(Web):2 FEB 2007
DOI:10.1002/adsc.200600355
The efficient tandem rhodium-catalysed 1,4-addition/cyclisation of 1,1′-alkenes using arylzinc chlorides is described. The simple one-step synthesis of substituted cyclopentanone and cyclohexanone derivatives is performed from acyclic precursors using relatively low catalyst loadings under mild conditions. A new quaternary carbon centre is created during the cyclisation step.
Co-reporter:Jonathan D. Hargrave, Gerwyn Bish and Christopher G. Frost
Chemical Communications 2006 (Issue 42) pp:4389-4391
Publication Date(Web):03 Oct 2006
DOI:10.1039/B610128C
The appropriate choice of organometallic nucleophile enables the straightforward preparation of different stereoisomers of 2-substituted pyrrolizidinones utilising the rhodium-catalysed 1,4-addition reaction.
Co-reporter:Thomas M. Douglas, Jérôme Le Nôtre, Simon K. Brayshaw, Christopher G. Frost and Andrew S. Weller
Chemical Communications 2006 (Issue 32) pp:3408-3410
Publication Date(Web):21 Jul 2006
DOI:10.1039/B608129K
Facile, metal-mediated, (acceptorless) dehydrogenation of tricyclopentyl phosphine directly affords rhodium chelating phosphine–olefin complexes, some of which are catalytically active for 1,4-additions.
Co-reporter:Jonathan D. Hargrave, Jennifer Herbert, Gerwyn Bish and Christopher G. Frost
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 17) pp:3235-3241
Publication Date(Web):26 Jul 2006
DOI:10.1039/B606977K
The cationic rhodium complex [Rh(cod)2][BF4] effectively catalyses the 1,4-addition of organotrialkoxysilanes to α-substituted acrylic esters. The reactions are promoted by heating in an oil-bath or more conveniently in a microwave reactor allowing rapid access to a useful range of functionalised products including 2-alkyl succinates and α-amino acid derivatives.
Co-reporter:Rebecca J. Moss, Kelly J. Wadsworth, Christopher J. Chapman and Christopher G. Frost
Chemical Communications 2004 (Issue 17) pp:1984-1985
Publication Date(Web):08 Jul 2004
DOI:10.1039/B406905F
The rhodium catalysed addition of potassium trifluoro(organo)borates to dimethyl itaconate generates an intermediate complex which on protonation provides enantioenriched succinic esters.
Co-reporter:Christopher J. Chapman;Christopher G. Frost
Advanced Synthesis & Catalysis 2003 Volume 345(Issue 3) pp:
Publication Date(Web):7 MAR 2003
DOI:10.1002/adsc.200390039
The efficient synthesis of substituted phenylalanine-type amino acids using a rhodium-catalysed, conjugate addition of arylboronic acids is described. The reactions are run in water and use a low loading (0.5 mol %) of rhodium catalyst.
Co-reporter:Nathan J. Patmore;Catherine Hague;Jamie H. Cotgreave;Mary F. Mahon Dr. Dr.;Andrew S. Weller Dr.
Chemistry - A European Journal 2002 Volume 8(Issue 9) pp:
Publication Date(Web):24 APR 2002
DOI:10.1002/1521-3765(20020503)8:9<2088::AID-CHEM2088>3.0.CO;2-L
Four Lewis acidic silver phosphane complexes partnered with [1-closo-CB11H12]− and [1-closo-CB11H6Br6]− have been synthesised and studied by solution NMR and solid-state X-ray diffraction techniques. In the complex [Ag(PPh3)(CB11H12)] (1), the silver is coordinated with the carborane by two stronger 3c–2e B−H−Ag bonds, one weaker B−H−Ag interaction and a very weak Ag⋅⋅⋅Carene contact in the solid state. In solution, the carborane remains closely connected with the {Ag(PPh3)}+ fragment, as evidenced by 11B chemical shifts. Complex 2 [Ag(PPh3)2(CB11H12)]2 adopts a dimeric motif in the solid state, each carborane bridging two Ag centres. In solution at low temperature, two distinct complexes are observed that are suggested to be monomeric [Ag(PPh3)2][CB11H12] and dimeric [Ag(PPh3)2(CB11H12)]2. With the more weakly coordinating anion [CB11H6Br6]− and one phosphane, complex 3 [Ag(PPh3)(CB11H6Br6)] is isolated. Complex 4, [Ag(PPh3)2(CB11H6Br6)], has been characterised spectroscopically. All of the complexes have been assessed as Lewis acids in the hetero-Diels–Alder reaction of N-benzylideneaniline with Danishefsky's diene. Exceptionally low catalyst loadings for this Lewis acid catalysed reaction are required (0.1 mol %) coupled with turnover frequencies of 4000 h−1 (quantitative conversion to product after 15 minutes using 3 at room temperature). Moreover, the reaction does not occur in rigorously dry solvent as addition of a substoichiometric amount of water (50 mol %) is necessary for turnover of the catalyst. It is suggested that a Lewis assisted Brønsted acid is formed between the water and the silver. The effect of changing the counterion to [BF4]−, [OTf]− and [ClO4]− has also been studied. Significant decreases in reaction rate and final product yield are observed on changing the anion from [CB11H6Br6]−, thus demonstrating the utility of weakly coordinating carborane anions in organic synthesis.
Co-reporter:Christopher G. Frost and Kelly J. Wadsworth
Chemical Communications 2001 (Issue 22) pp:2316-2317
Publication Date(Web):24 Oct 2001
DOI:10.1039/B107633G
The efficient transmetalation from boron to rhodium is exploited in a new synthesis of aryl and alkenyl ketones.
Co-reporter:Catherine Hague, Nathan J. Patmore, Christopher G. Frost, Mary F. Mahon and Andrew S. Weller
Chemical Communications 2001 (Issue 21) pp:2286-2287
Publication Date(Web):17 Oct 2001
DOI:10.1039/B106719B
The complex [(PPh3)Ag(CB11H6Br6)] 1 is an effective and selective catalyst (0.1 mol% loading) for a hetero-Diels–Alder reaction, which shows a marked dependence on the presence of trace amounts of water, while addition of Ag[Y] [Y = CB11H12, CB11H6Br6, O3SCF3] to a phosphine functionalised support gives an efficient and recyclable Lewis acid catalyst for this transformation.
Co-reporter:Christopher G Frost, Paul Mendonça
Tetrahedron: Asymmetry 2000 Volume 11(Issue 9) pp:1845-1848
Publication Date(Web):19 May 2000
DOI:10.1016/S0957-4166(00)00150-6
The rational modification of an established amino-alcohol scaffold has revealed new, highly effective ligands for the enantioselective transfer hydrogenation of acetophenone that affords the product in up to 95% ee.
Co-reporter:Hannah J. Edwards, Jonathan D. Hargrave, Stephen D. Penrose and Christopher G. Frost
Chemical Society Reviews 2010 - vol. 39(Issue 6) pp:NaN2105-2105
Publication Date(Web):2010/04/21
DOI:10.1039/B919762C
The rhodium catalysed conjugate addition of organometallics to activated alkenes is a powerful synthetic tool for establishing new carbon–carbon bonds often with high stereoselectivity. The introduction of a practical, efficient method for introducing functionalised aryl and alkenyl fragments with predictable stereocontrol has caught the attention of synthetic chemists and emerging examples are growing in number and complexity. In this tutorial review, we document notable advances in the application of rhodium catalysed conjugate addition processes within the context of synthesis of complex molecules and intermediates in drug discovery. The chosen examples illustrate important issues regarding scope, selectivity and reactivity that will help guide the selection of appropriate donor and acceptor to achieve the desired carbon–carbon bond construction when planning new synthetic routes.
Co-reporter:Christopher J. Chapman, Ai Matsuno, Christopher G. Frost and Michael C. Willis
Chemical Communications 2007(Issue 38) pp:NaN3905-3905
Publication Date(Web):2007/08/15
DOI:10.1039/B711533D
The site-selective modification of peptides containing dehydroalanine, tyrosine and tryptophan residues has been achieved using rhodium catalysed conjugate additions or palladium catalysed aryl-amination and -etherification reactions.
Co-reporter:Jérôme Le Nôtre, Joseph C. Allen and Christopher G. Frost
Chemical Communications 2008(Issue 32) pp:NaN3797-3797
Publication Date(Web):2008/06/23
DOI:10.1039/B806098C
The enantioselective 1,4-addition of 2-(substituted)thienylzinc and 2-furanylzinc reagents has been achieved (up to 99 : 1 er) using a complex derived from [Rh(C2H4)2Cl]2 and Me-DUPHOS.
Co-reporter:Christopher G. Frost, Stephen D. Penrose and Robert Gleave
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 23) pp:NaN4347-4347
Publication Date(Web):2008/10/18
DOI:10.1039/B812897A
The concise enantioselective synthesis of hermitamides A and B is presented utilising a rhodium catalysed conjugate addition reaction to introduce the side chain and chiral information in a single step via an alkenyltrifluoroborate salt.
Co-reporter:Andrew J. Paterson, Callum J. Heron, Claire L. McMullin, Mary F. Mahon, Neil J. Press and Christopher G. Frost
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 28) pp:NaN6000-6000
Publication Date(Web):2017/06/26
DOI:10.1039/C7OB01192J
A catalytic meta selective C–H alkylation of arenes is described using a wide range of α-halo carbonyls as coupling partners. Previously unreported primary alkylations with high meta selectivity have been enabled by this methodology whereas using straight chain alkyl halides affords ortho substituted products. Mechanistic analysis reveals an activation pathway whereby cyclometalation with a ruthenium(II) complex activates the substrate molecule and is responsible for the meta selectivity observed. A distinct second activation of the coupling partner allows site selective reaction between both components.
Co-reporter:Patricia Marcé, Andrew J. Paterson, Mary F. Mahon and Christopher G. Frost
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 19) pp:NaN7076-7076
Publication Date(Web):2016/06/29
DOI:10.1039/C6CY01254J
The catalytic meta-functionalization of arenes has emerged as an important synthetic methodology in the last decade. We report herein structural and mechanistic studies of the meta-sulfonation of phenylpyridine using ruthenium complexes. Furthermore, we disclose that the catalytically active species does not require the presence of a η6-arene ligand. Furthermore, the novel cycloruthenated phenylpyridine complex tosylated at the para position to the metal has been isolated and fully characterised. Protodemetallation studies suggest that a concerted C–H activation–demetallation process may be involved. Overall, this study provides fundamental insight into the meta-sulfonation phenylpyridine reaction pathway and uncovers new reaction intermediates that will guide the design of new catalytic systems for remote meta-functionalization.
Co-reporter:Sean Goggins, Eleanor Rosevere, Clément Bellini, Joseph C. Allen, Barrie J. Marsh, Mary F. Mahon and Christopher G. Frost
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 1) pp:NaN52-52
Publication Date(Web):2013/11/07
DOI:10.1039/C3OB42099J
The synthesis of a range of novel silyl-protected dioxaborinanes as a column- and bench-stable boron reagent were found to be advantageous to achieving good yields in palladium-catalysed cross-coupling reactions under standard conditions.
Co-reporter:Joseph C. Allen, Gabriele Kociok-Köhn and Christopher G. Frost
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 1) pp:NaN35-35
Publication Date(Web):2011/10/05
DOI:10.1039/C1OB06586F
The rhodium-catalysed enantioselective 1,4-addition of organoboron reagents to arylidene Meldrum's acids as acceptors, allows convenient access to 4-arylchroman-2-ones with good to excellent levels of enantioselectivity. The use of silyl-protected dioxaborinanes as donors was found to be advantageous to achieving good yields of product under anhydrous conditions.
Co-reporter:Jonathan D. Hargrave, Gerwyn Bish, Gabriele Kociok Köhn and Christopher G. Frost
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 22) pp:NaN5125-5125
Publication Date(Web):2010/09/08
DOI:10.1039/C0OB00158A
The rhodium-catalysed conjugate addition of arylboronic acids to an enantiopure acceptor derived from (R)-S-methylcysteine proceeds under substrate control to provide a range of functionalised phenylalanine derivatives with excellent stereocontrol via a highly diastereoselective protonation.
Co-reporter:Sean Goggins, Christophe Naz, Barrie J. Marsh and Christopher G. Frost
Chemical Communications 2015 - vol. 51(Issue 3) pp:NaN564-564
Publication Date(Web):2014/11/10
DOI:10.1039/C4CC07693A
A novel ferrocene-derived substrate for the ratiometric electrochemical detection of alkaline phosphatase (ALP) was designed and synthesised. It was demonstrated to be an excellent electrochemical substrate for the ALP-labelled enzyme-linked immunosorbent assay (ELISA).
Co-reporter:Sean Goggins, Barrie J. Marsh, Anneke T. Lubben and Christopher G. Frost
Chemical Science (2010-Present) 2015 - vol. 6(Issue 8) pp:NaN4985-4985
Publication Date(Web):2015/06/15
DOI:10.1039/C5SC01588J
Signal transduction and signal amplification are both important mechanisms used within biological signalling pathways. Inspired by this process, we have developed a signal amplification methodology that utilises the selectivity and high activity of enzymes in combination with the robustness and generality of an organometallic catalyst, achieving a hybrid biological and synthetic catalyst cascade. A proligand enzyme substrate was designed to selectively self-immolate in the presence of the enzyme to release a ligand that can bind to a metal pre-catalyst and accelerate the rate of a transfer hydrogenation reaction. Enzyme-triggered catalytic signal amplification was then applied to a range of catalyst substrates demonstrating that signal amplification and signal transduction can both be achieved through this methodology.
Co-reporter:William R. Reynolds, Pawel Plucinski and Christopher G. Frost
Catalysis Science & Technology (2011-Present) 2014 - vol. 4(Issue 4) pp:NaN954-954
Publication Date(Web):2014/01/23
DOI:10.1039/C3CY00836C
Two novel polymer encapsulated silica supported palladium catalysts have been prepared and shown to be highly active, robust and recyclable catalysts for Heck and Suzuki–Miyaura cross coupling reactions under continuous flow conditions. They were demonstrated to give excellent conversions of both electron rich and electron deficient substrates and the catalysts were used for over 50 hours continuous operation without any appreciable decrease in catalytic activity and low levels of Pd leaching measured by ICP-MS.
Co-reporter:Andrew J. Paterson, Sahra St John-Campbell, Mary F. Mahon, Neil J. Press and Christopher G. Frost
Chemical Communications 2015 - vol. 51(Issue 64) pp:NaN12810-12810
Publication Date(Web):2015/07/06
DOI:10.1039/C5CC03951G
A catalytic meta-selective C–H functionalization of 2-phenylpyridines using a range of tertiary halides is described. The protocol is simple to perform and uses commercially available reagents to construct challenging quaternary carbon centres in a regioselective manner. Preliminary studies suggest the C–H functionalization proceeds through a radical process directed via a remote σ-activation.



![L-Serine, N-[N-[(1,1-dimethylethoxy)carbonyl]-L-tryptophyl]-, methyl ester](http://img.cochemist.com/ccimg/81200/81187-76-0.png)
![L-Serine, N-[N-[(1,1-dimethylethoxy)carbonyl]-L-tryptophyl]-, methyl ester](http://img.cochemist.com/ccimg/81200/81187-76-0_b.png)
![Glycine, N-[(1,1-dimethylethoxy)carbonyl]-L-tryptophyl-, ethyl ester](http://img.cochemist.com/ccimg/38500/38428-01-2.png)
![Glycine, N-[(1,1-dimethylethoxy)carbonyl]-L-tryptophyl-, ethyl ester](http://img.cochemist.com/ccimg/38500/38428-01-2_b.png)
![1H-Pyrazole, 1-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/35800/35715-68-5.png)
![1H-Pyrazole, 1-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/35800/35715-68-5_b.png)
![Silane, trimethoxy[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/35700/35692-32-1.png)
![Silane, trimethoxy[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/35700/35692-32-1_b.png)


![2-Propenamide, N-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/25700/25681-32-7.png)
![2-Propenamide, N-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/25700/25681-32-7_b.png)





![BUTANEDIOIC ACID, [(4-METHOXYPHENYL)METHYL]-, DIMETHYL ESTER](http://img.cochemist.com/ccimg/793000/792942-87-1.png)
![BUTANEDIOIC ACID, [(4-METHOXYPHENYL)METHYL]-, DIMETHYL ESTER](http://img.cochemist.com/ccimg/793000/792942-87-1_b.png)
![PYRIDINE, 3-[(1E)-2-PHENYLETHENYL]-](http://img.cochemist.com/ccimg/5100/5097-91-6.png)
![PYRIDINE, 3-[(1E)-2-PHENYLETHENYL]-](http://img.cochemist.com/ccimg/5100/5097-91-6_b.png)

![L-Serine, N-[N-[(1,1-dimethylethoxy)carbonyl]-L-tyrosyl]-, methyl ester](http://img.cochemist.com/ccimg/16900/16874-54-7.png)
![L-Serine, N-[N-[(1,1-dimethylethoxy)carbonyl]-L-tyrosyl]-, methyl ester](http://img.cochemist.com/ccimg/16900/16874-54-7_b.png)
![(1R,4R,8R)-5-Benzyl-2-isobutyl-8-methoxy-1,8-dimethylbicyclo[2.2.2]octa-2,5 -diene](http://img.cochemist.com/ccimg/948600/948594-95-4.png)
![(1R,4R,8R)-5-Benzyl-2-isobutyl-8-methoxy-1,8-dimethylbicyclo[2.2.2]octa-2,5 -diene](http://img.cochemist.com/ccimg/948600/948594-95-4_b.png)






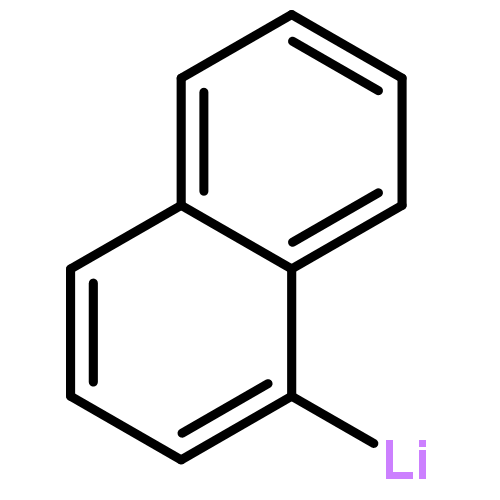
![2-[(1-naphthylmethyl)amino]ethanol](http://img.cochemist.com/ccimg/20200/20103-08-6.png)
![2-[(1-naphthylmethyl)amino]ethanol](http://img.cochemist.com/ccimg/20200/20103-08-6_b.png)










![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-valyl-, methyl ester](/data/chemimg/1786900/20902-47-0.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-valyl-, methyl ester](/data/chemimg/1786900/20902-47-0_b.png)
![Ethanone, 1-[4-[(1E)-2-phenylethenyl]phenyl]-](/data/chemimg/1785200/20488-42-0.png)
![Ethanone, 1-[4-[(1E)-2-phenylethenyl]phenyl]-](/data/chemimg/1785200/20488-42-0_b.png)
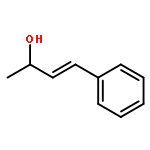
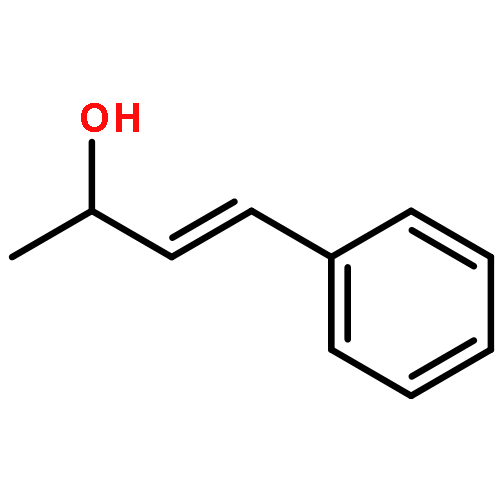


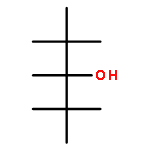
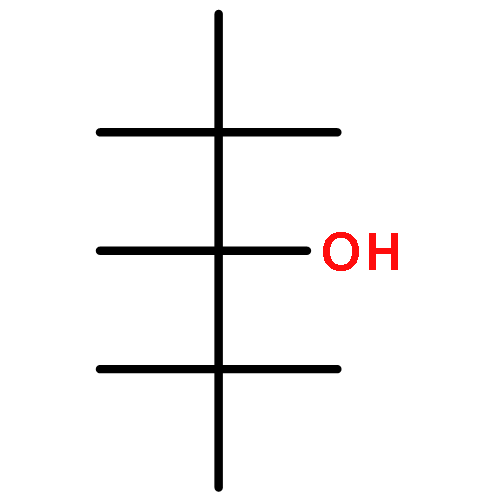


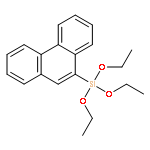
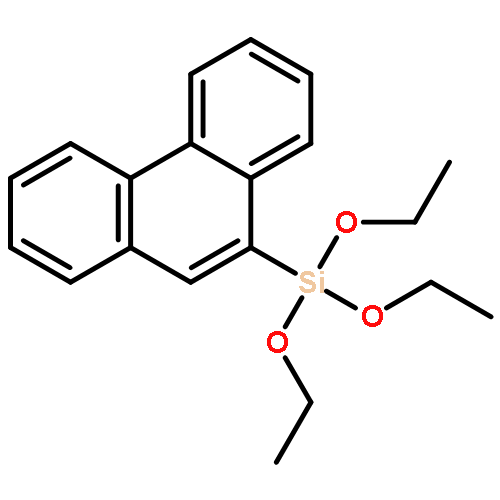
![1,3-Dioxane-4,6-dione, 5-[(4-methoxyphenyl)methylene]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15800/15795-54-7.png)
![1,3-Dioxane-4,6-dione, 5-[(4-methoxyphenyl)methylene]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15800/15795-54-7_b.png)
![[1,1'-Biphenyl]-4-yltriethoxysilane](http://img.cochemist.com/ccimg/18100/18056-97-8.png)
![[1,1'-Biphenyl]-4-yltriethoxysilane](http://img.cochemist.com/ccimg/18100/18056-97-8_b.png)
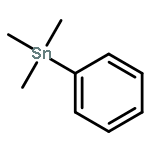
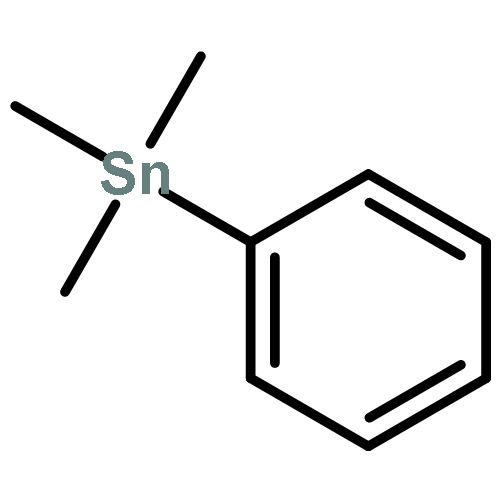
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)


![Chromium, [(2,3,5,6-h)-bicyclo[2.2.1]hepta-2,5-diene]tetracarbonyl-](http://img.cochemist.com/ccimg/12200/12146-36-0.png)
![Chromium, [(2,3,5,6-h)-bicyclo[2.2.1]hepta-2,5-diene]tetracarbonyl-](http://img.cochemist.com/ccimg/12200/12146-36-0_b.png)


![1,3-Dioxane-4,6-dione, 2,2-dimethyl-5-[(4-nitrophenyl)methylene]-](http://img.cochemist.com/ccimg/15800/15795-62-7.png)
![1,3-Dioxane-4,6-dione, 2,2-dimethyl-5-[(4-nitrophenyl)methylene]-](http://img.cochemist.com/ccimg/15800/15795-62-7_b.png)
![Benzenemethanol, α-[(trimethylsilyl)methyl]-](/data/chemimg/1762100/17993-97-4.png)
![Benzenemethanol, α-[(trimethylsilyl)methyl]-](/data/chemimg/1762100/17993-97-4_b.png)
![1,3-Dioxane-4,6-dione, 5-[(2-methoxyphenyl)methylene]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15800/15795-56-9.png)
![1,3-Dioxane-4,6-dione, 5-[(2-methoxyphenyl)methylene]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15800/15795-56-9_b.png)


![1-(4'-Methoxy-[1,1'-biphenyl]-4-yl)ethanone](http://img.cochemist.com/ccimg/13100/13021-18-6.png)
![1-(4'-Methoxy-[1,1'-biphenyl]-4-yl)ethanone](http://img.cochemist.com/ccimg/13100/13021-18-6_b.png)


![Benzene, 1-methoxy-4-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/1300/1216-95-1.png)
![Benzene, 1-methoxy-4-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/1300/1216-95-1_b.png)






![1-chloro-4-[(4-methylphenyl)sulfonyl]benzene](http://img.cochemist.com/ccimg/5200/5184-71-4.png)
![1-chloro-4-[(4-methylphenyl)sulfonyl]benzene](http://img.cochemist.com/ccimg/5200/5184-71-4_b.png)

![Benzene,1,3,5-trimethyl-2-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/5200/5184-64-5.png)
![Benzene,1,3,5-trimethyl-2-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/5200/5184-64-5_b.png)
![Bicyclo[2.2.2]octa-2,5-diene,8-methoxy-1,8-dimethyl-2-(2-methylpropyl)-5-(phenylmethyl)-, (1S,4S,8S)-](http://img.cochemist.com/ccimg/862500/862499-50-1.png)
![Bicyclo[2.2.2]octa-2,5-diene,8-methoxy-1,8-dimethyl-2-(2-methylpropyl)-5-(phenylmethyl)-, (1S,4S,8S)-](http://img.cochemist.com/ccimg/862500/862499-50-1_b.png)






![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0.png)
![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0_b.png)
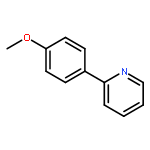
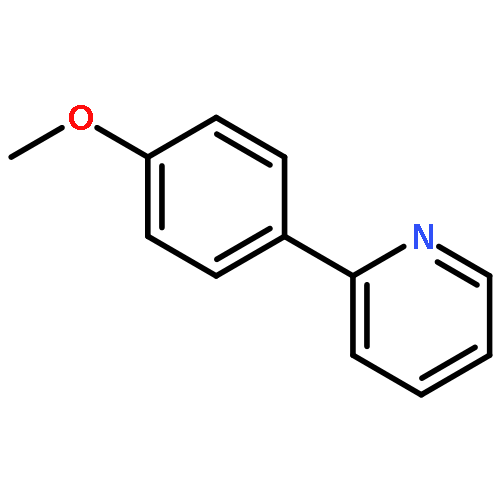
![2-[(e)-2-phenylethenyl]pyridine](http://img.cochemist.com/ccimg/600/538-49-8.png)
![2-[(e)-2-phenylethenyl]pyridine](http://img.cochemist.com/ccimg/600/538-49-8_b.png)















![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0.png)
![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0_b.png)


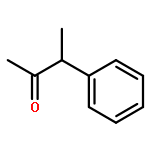
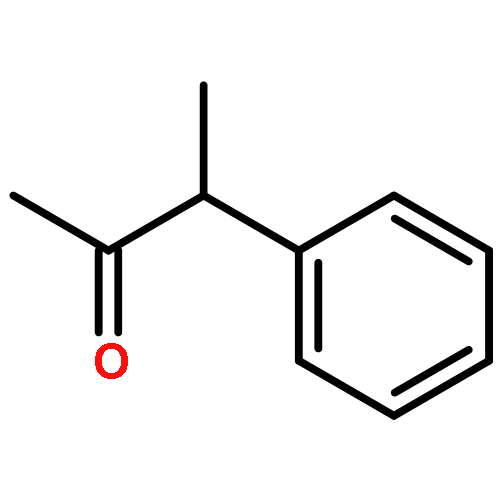




![Benzene, 2,4-dimethyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/13300/13249-97-3.png)
![Benzene, 2,4-dimethyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/13300/13249-97-3_b.png)
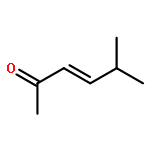
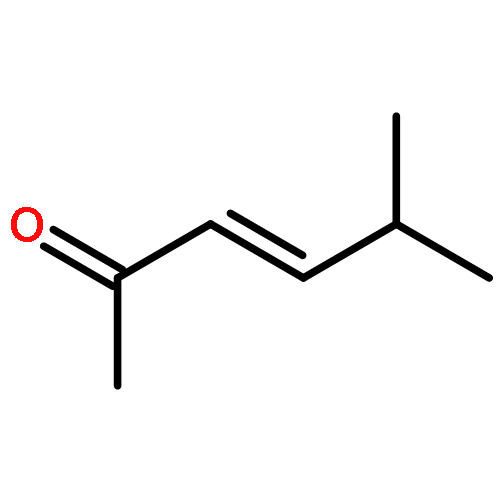
![Benzene,1-fluoro-4-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/1700/1643-97-6.png)
![Benzene,1-fluoro-4-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/1700/1643-97-6_b.png)

























![1,3-Dioxane-4,6-dione,5-[[4-(dimethylamino)phenyl]methylene]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15800/15795-57-0.png)
![1,3-Dioxane-4,6-dione,5-[[4-(dimethylamino)phenyl]methylene]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15800/15795-57-0_b.png)
![Methyl 2-[[2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-phenylpropanoyl]amino]-3-phenylpropanoate](http://img.cochemist.com/ccimg/15300/15215-74-4.png)
![Methyl 2-[[2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-phenylpropanoyl]amino]-3-phenylpropanoate](http://img.cochemist.com/ccimg/15300/15215-74-4_b.png)










![1,3-Dioxane-4,6-dione, 5-[(4-methoxyphenyl)methyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/62000/61958-46-1.png)
![1,3-Dioxane-4,6-dione, 5-[(4-methoxyphenyl)methyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/62000/61958-46-1_b.png)


![Benzene, 1-bromo-4-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/5200/5184-70-3.png)
![Benzene, 1-bromo-4-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/5200/5184-70-3_b.png)

















![3-(acetyloxy)-2-[(acetyloxy)methyl]-2-methylpropyl acetate](http://img.cochemist.com/ccimg/13500/13431-59-9.png)
![3-(acetyloxy)-2-[(acetyloxy)methyl]-2-methylpropyl acetate](http://img.cochemist.com/ccimg/13500/13431-59-9_b.png)






















![1-[(4-methylphenyl)sulfonyl]piperidine](http://img.cochemist.com/ccimg/4800/4703-22-4.png)
![1-[(4-methylphenyl)sulfonyl]piperidine](http://img.cochemist.com/ccimg/4800/4703-22-4_b.png)








![Acetamide,N-[1,1'-biphenyl]-2-yl-](http://img.cochemist.com/ccimg/2200/2113-47-5.png)
![Acetamide,N-[1,1'-biphenyl]-2-yl-](http://img.cochemist.com/ccimg/2200/2113-47-5_b.png)
![Ethanone,1-[1,1'-biphenyl]-3-yl-](http://img.cochemist.com/ccimg/3200/3112-01-4.png)
![Ethanone,1-[1,1'-biphenyl]-3-yl-](http://img.cochemist.com/ccimg/3200/3112-01-4_b.png)








![Ethanone,1-[1,1'-biphenyl]-2-yl-](http://img.cochemist.com/ccimg/2200/2142-66-7.png)
![Ethanone,1-[1,1'-biphenyl]-2-yl-](http://img.cochemist.com/ccimg/2200/2142-66-7_b.png)
![2-phenyltetrahydro-1H-pyrrolo[1,2-c]imidazole-1,3(2H)-dione](http://img.cochemist.com/ccimg/2300/2221-09-2.png)
![2-phenyltetrahydro-1H-pyrrolo[1,2-c]imidazole-1,3(2H)-dione](http://img.cochemist.com/ccimg/2300/2221-09-2_b.png)