Co-reporter:Stephen H. Frayne, Raghavendra R. Murthy, and Brian H. Northrop
The Journal of Organic Chemistry August 4, 2017 Volume 82(Issue 15) pp:7946-7946
Publication Date(Web):July 11, 2017
DOI:10.1021/acs.joc.7b01200
Thiol-Michael “click” reactions are essential synthetic tools in the preparation of various materials including polymers, dendrimers, and other macromolecules. Despite increasing efforts to apply thiol-Michael chemistry in a controlled fashion, the selectivity of base- or nucleophile-promoted thiol-Michael reactions in complex mixtures of multiple thiols and/or acceptors remains largely unknown. Herein, we report a thorough fundamental study of the selectivity of thiol-Michael reactions through a series of 270 ternary reactions using 1H NMR spectroscopy to quantify product selectivity. The varying influences of different catalysts/initiators are explored using ternary reactions between two Michael acceptors and a single thiol or between a single Michael acceptor and two thiols using three different catalysts/initiators (triethylamine, DBU, and dimethylphenylphosphine) in chloroform. The results from the ternary reactions provide a platform from which sequential quaternary, one-pot quaternary, and sequential senary thiol-Michael reactions were designed and their selectivities quantified. These results provide insights into the design of selective thiol-Michael reactions that can be used for the synthesis and functionalization of multicomponent polymers and further informs how catalyst/initiator choice influences the reactivity between a given thiol and Michael acceptor.
Co-reporter:Anton D. Chavez, Brian J. Smith, Merry K. Smith, Peter A. Beaucage, Brian H. Northrop, and William R. Dichtel
Chemistry of Materials 2016 Volume 28(Issue 14) pp:4884
Publication Date(Web):June 24, 2016
DOI:10.1021/acs.chemmater.6b01831
Co-reporter:Alexander R. Goldberg and Brian H. Northrop
The Journal of Organic Chemistry 2016 Volume 81(Issue 3) pp:969-980
Publication Date(Web):January 6, 2016
DOI:10.1021/acs.joc.5b02548
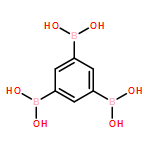
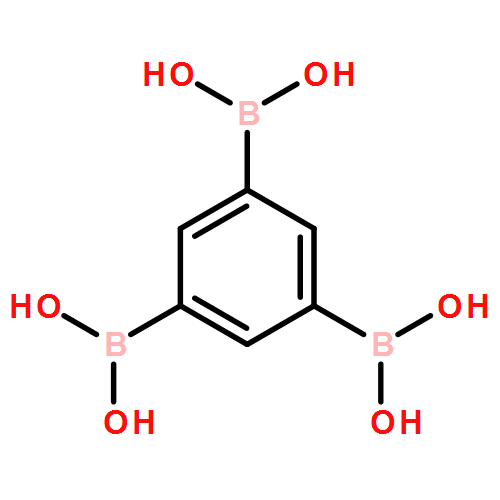
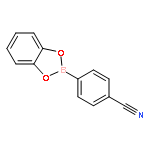
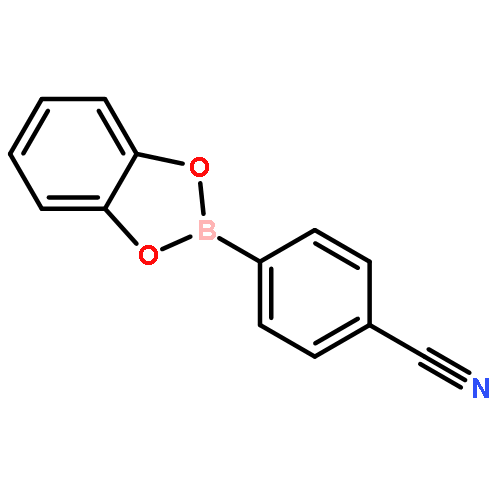
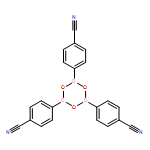
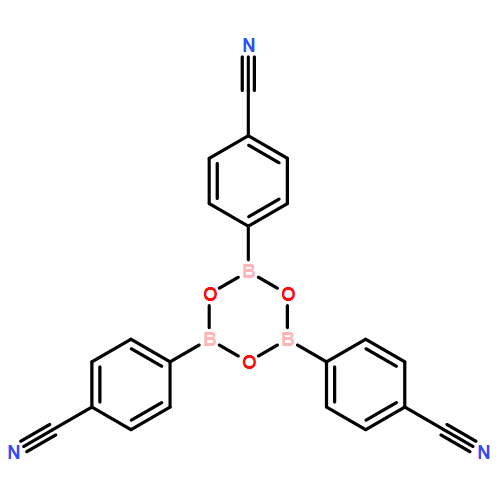
The solution phase self-assembly of boronate esters, diazaboroles, oxathiaboroles, and dithiaboroles from the condensation of arylboronic acids with aromatic diol, diamine, hydroxythiol, and dithiol compounds in chloroform has been investigated by 1H NMR spectroscopy and computational methods. Six arylboronic acids were included in the investigations with each boronic acid varying in the substituent at its 4-position. Both computational and experimental results show that the para-substituent of the arylboronic acid does not significantly influence the favorability of forming a condensation product with a given organic donor. The type of donor, however, greatly influences the favorability of self-assembly. 1H NMR spectroscopy indicates that condensation reactions between arylboronic acids and catechol to give boronate esters are the most favored thermodynamically, followed by diazaborole formation. Computational investigations support this conclusion. Neither oxathiaboroles nor dithiaboroles form spontaneously at equilibrium in chloroform at room temperature. Computational results suggest that the effect of borylation on the frontier orbitals of each donor helps to explain differences in the favorability of their condensation reactions with arylboronic acids. The results can inform the use of boronic acids as they are increasingly utilized in the dynamic self-assembly of organic materials and as components in dynamic combinatorial libraries.
Co-reporter:Brian H. Northrop, Stephen H. Frayne and Umesh Choudhary
Polymer Chemistry 2015 vol. 6(Issue 18) pp:3415-3430
Publication Date(Web):23 Mar 2015
DOI:10.1039/C5PY00168D
The mechanism and kinetics of thiol–maleimide “click” reactions carried out under a variety of conditions have been investigated computationally and using experimental competition reactions. The influence of three different solvents (chloroform, ethane thiol, and N,N-dimethylformamide), five different initiators (ethylamine, diethylamine, triethylamine, diazabicyclo[2.2.2]octane, and dimethylphenyl-phosphine), and seven different thiols (methyl mercaptan, β-mercaptoethanol, thioacetic acid, methyl thioglycolate, methyl 3-mercaptopropionate, cysteine methyl ester, and thiophenol) on the energetics and kinetics of thiol–maleimide reactions have been examined using density functional methods. Computational and kinetic modeling indicate that the choice of solvent, initiator, and thiol directly influences whether product formation follows a base-, nucleophile-, or ion pair-initiated mechanism (or some combination thereof). The type of mechanism followed determines the overall thiol–maleimide reaction kinetics. Insights from computational studies are then used to understand the selectivity of ternary thiol–maleimide reactions between N-methyl maleimide, thiophenol, and 1-hexanethiol in different combinations of solvents and initiators. The results provide considerable insight into the interplay between reaction conditions, kinetics, and selectivity in thiol–maleimide reactions in particular and thiol-Michael reactions in general, with implications ranging from small molecule synthesis to bioconjugation chemistry and multifunctional materials.
Co-reporter:Merry K. Smith;Alexer R. Goldberg ;Brian H. Northrop
European Journal of Organic Chemistry 2015 Volume 2015( Issue 13) pp:2928-2941
Publication Date(Web):
DOI:10.1002/ejoc.201500170
Abstract
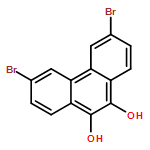
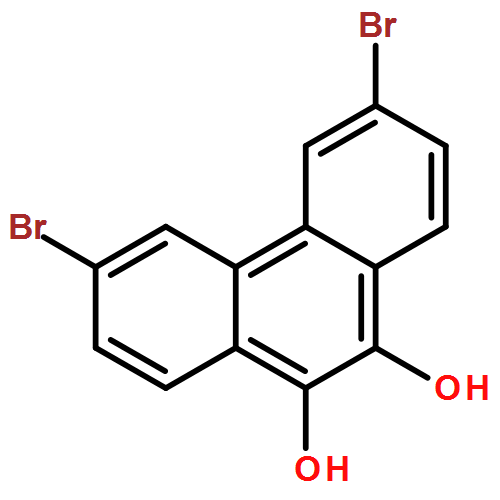
Six variably functionalized phenanthrene-based bis(catechol) derivatives have been synthesized and their ability to undergo dynamic covalent assembly with 1,4-benzenediboronic acid (BDBA) to give discrete, shape-persistent [2+2] assemblies has been investigated. Systematically varying the functionality of the bis(catechol) derivatives is found to influence both the reaction conditions necessary to promote their self-assembly with BDBA as well as the stability of the resulting assemblies in the presence of protic solvents. A model is proposed to describe how solvent choice and starting material functionality must be carefully balanced in order to shift many competing equilibria toward the formation of discrete, soluble covalent organic polygons. The results are expected to provide additional insight into optimizing the synthesis of other boronate ester polygons/polyhedra as well as related, “infinite” covalent organic frameworks (COFs).
Co-reporter:Merry K. Smith; Samantha R. Angle;Brian H. Northrop
Journal of Chemical Education 2015 Volume 92(Issue 2) pp:368-372
Publication Date(Web):November 6, 2014
DOI:10.1021/ed500540t
γ-Cyclodextrin can assemble in the presence of KOH or RbOH into metal–organic frameworks (CD–MOFs) with applications in gas adsorption and environmental remediation. Crystalline CD–MOFs are grown by vapor diffusion and their reversible adsorption of CO2(g) is analyzed both qualitatively and quantitatively. The experiment can be tailored to high school through advanced undergraduate laboratories and engages students in several areas of fundamental chemistry (crystallization, chemical equilibria, acid–base reactions, gas laws), advanced materials (MOFs), and broader impacts of chemistry (renewable resources and environmental chemistry).
Co-reporter:Merry K. Smith and Brian H. Northrop
Chemistry of Materials 2014 Volume 26(Issue 12) pp:3781
Publication Date(Web):May 29, 2014
DOI:10.1021/cm5013679
The vibrational characteristics of 28 different boronic acid, boroxine anhydride, and boronate ester species have been systematically investigated using a combination of experimental infrared (IR) spectroscopy and computational modeling. IR bands characteristic to each boron-containing functionality have been categorized and assigned in conjunction with density functional theory (B3LYP/6-31G(d)), with the aim of better understanding and distinguishing the vibrational characteristics of covalent organic frameworks (COFs) built from boronic acids. In several cases, vibrational assignments differ from those previously reported in the literature on boronic acid-based COFs. Vibrations commonly regarded as diagnostic for one functionality are found in regions of the IR spectrum where other functionalities also show characteristic peaks. The collective experimental and computational results reveal that several alternative bands in the IR region can be used to more diagnostically distinguish between boronic acid, boroxine anhydride, and boronate ester species. The results presented herein provide the tools for straightforward characterization of boroxine anhydride and boronate ester species using IR spectroscopy. The results can be applied to additional theoretical studies of larger COF-like assemblies as well as the analysis of other boronic-acid-based materials.
Co-reporter:Merry K. Smith, Natalia E. Powers-Riggs and Brian H. Northrop
RSC Advances 2014 vol. 4(Issue 72) pp:38281-38292
Publication Date(Web):15 Aug 2014
DOI:10.1039/C4RA06503D
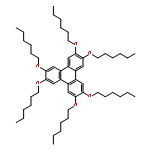
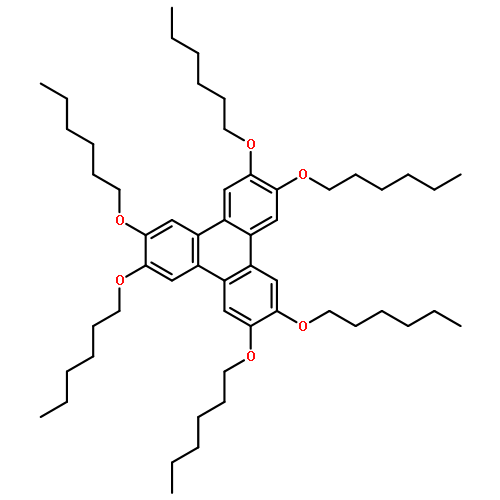
A straightforward, reliable, and scalable synthesis of rationally designed, mixed-substituent triphenylene derivatives from ortho-terphenyl precursors is described. Three isomers of bis(hexyloxy)-tetrahydroxy triphenylenes were synthesized and functionalized with monomethyl di(ethylene glycol) chains to provide new amphiphilic, mixed substituent triphenylenes. Oxidative triphenylene annulation, tetra-ol formation, and subsequent functionalization were supported by significant changes in phase and melting point, and confirmed by mass spectrometry, differential scanning calorimetry, and UV/Vis, 1H, and 13C NMR spectroscopies. The thermal phase properties of amphiphilic mixed-substituent triphenylene derivatives were found to vary between the different isomers, demonstrating how small changes in substitution pattern can result in significant differences in mesogenic behavior. The controlled synthetic route to de novo designed triphenylene derivatives is dependable, wide in scope, and can be applied to the synthesis of a vast array of other mixed-substituent triphenylene derivatives, thus enabling the preparation of libraries of novel triphenylene and triphenylene-containing materials.
Co-reporter:Umesh Choudhary ; Brian H. Northrop
Chemistry - A European Journal 2014 Volume 20( Issue 4) pp:999-1009
Publication Date(Web):
DOI:10.1002/chem.201303864
Abstract
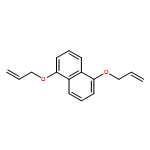
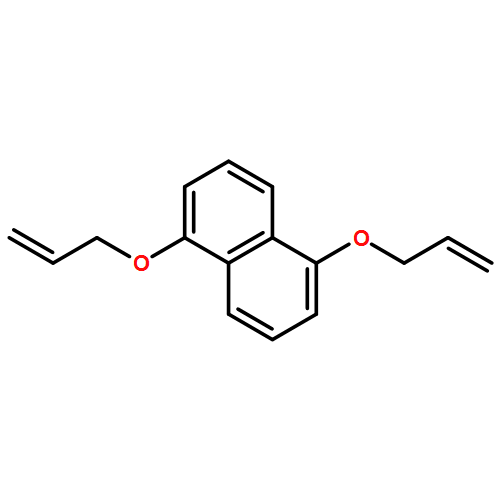
Five dioxynaphthalene[38]-crown-10 (DNP38C10) macrocycles bearing one, two, three, or four allyl moieties have been synthesized and their ability to spontaneously self-assemble with methyl viologen to form [2]pseudorotaxanes has been evaluated. Association constants between methyl viologen and several of the allyl-functionalized DNP38C10 macrocycles are found to be comparable to that of methyl viologen and unfunctionalized DNP38C10, however, the enthalpic and entropic factors that underlie overall binding free energy vary systematically with increasing allyl substitution. These variations are explained through a combination of solution phase and solid-state analysis of the macrocycles and their complexes. The utility of endowing DNP38C10 macrocycles with allyl moieties is further demonstrated by the ease with which they can be functionalized through thiol-ene click chemistry.
Co-reporter:Merry K. Smith, Natalia E. Powers-Riggs and Brian H. Northrop
Chemical Communications 2013 vol. 49(Issue 55) pp:6167-6169
Publication Date(Web):28 May 2013
DOI:10.1039/C3CC42777C
The facile self-assembly of nanoscale boronate ester rectangles from linear bis-catechols and 1,4-benzene diboronic acid is described. Spectroscopic and computational analyses reveal the influence of extended π-conjugation on the rectangles' absorption and fluorescence properties. The rectangles represent a new class of discrete, organic soluble covalent organic polygons.
Co-reporter:Robert M. Stolz and Brian H. Northrop
The Journal of Organic Chemistry 2013 Volume 78(Issue 16) pp:8105-8116
Publication Date(Web):July 31, 2013
DOI:10.1021/jo4014436
A combination of experimental and computational methods has been used to understand the reactivity and selectivity of orthogonal thiol–ene and thiol–yne ″click″ reactions involving N-allyl maleimide (1) and N-propargyl maleimide (2). Representative thiols methyl-3-mercaptopropionate and β-mercaptoethanol are shown to add exclusively and quantitatively to the electron poor maleimide alkene of 1 and 2 under base (Et3N) initiated thiol-Michael conditions. Subsequent radical-mediated thiol–ene or thiol–yne reactions can be carried out to further functionalize the remaining allyl or propargyl moieties in near quantitative yields (>95%). Selectivity, however, can only be achieved when base-initiated thiol-Michael reactions are carried out first, as radical-mediated reactions between equimolar amounts of thiol and N-substituted maleimides give complex mixtures of products. CBS-QB3 calculations have been used to investigate the energetics and kinetics of reactions between a representative thiol (methyl mercaptan) with N-allyl and N-propargyl maleimide under both base-initiated and radical-mediated conditions. Calculations help elucidate the factors that underlie the selective base-initiated and nonselective radical-mediated thiol–ene/yne reactions. The results provide additional insights into how to design selective radical-mediated thiol–ene/yne reactions.
Co-reporter:Umesh Choudhary and Brian H. Northrop
Organic Letters 2012 Volume 14(Issue 8) pp:2082-2085
Publication Date(Web):April 6, 2012
DOI:10.1021/ol300614z
Base-catalyzed thiol-maleimide click chemistry has been applied to the synthesis of neutral donor–acceptor [2]rotaxanes in good yield. This method is extended further to the synthesis of a glutathione-functionalized [2]pseudorotaxane, a precursor to integrated conjugates of interlocked molecules with proteins and enzymes.
Co-reporter:Robert C. Boutelle and Brian H. Northrop
The Journal of Organic Chemistry 2011 Volume 76(Issue 19) pp:7994-8002
Publication Date(Web):August 26, 2011
DOI:10.1021/jo201606z
The effects of furan and maleimide substitution on the dynamic reversibility of their Diels–Alder reactivity have been investigated computationally and by 1H NMR spectroscopy. Furan and furan derivatives bearing methoxy, methyl, or formyl groups at their 2- or 3-positions were investigated with maleimide and maleimide derivatives bearing N-methyl, N-allyl, and N-phenyl substituents. Computational predictions indicate that electronic and regiochemical effects of furan substitution significantly influence their Diels–Alder reactivity with maleimide, with reaction free energies of exo adduct formation ranging from ΔG = −9.4 to 0.9 kcal/mol and transition state barriers to exo adduct formation ranging from ΔG⧧ = 18.9 to 25.6 kcal/mol. Much less variation was observed for the reactivity of N-substituted maleimide derivatives and furan, with reaction and transition state free energies each falling within a range of 1.1 kcal/mol. Dynamic exchange experiments monitored by 1H NMR spectroscopy support computational predictions. The results indicate the reactivity and reversibility of furan–maleimide cycloadditions can be tuned significantly through the addition of appropriate substituents and have implications in the use of furan and maleimide derivatives in the construction of thermally responsive organic materials.
Co-reporter:Brian H. Northrop ;Roderick N. Coffey
Journal of the American Chemical Society () pp:
Publication Date(Web):August 1, 2012
DOI:10.1021/ja305441d
The influence of alkene functionality on the energetics and kinetics of radical initiated thiol–ene click chemistry has been studied computationally at the CBS-QB3 level. Relative energetics (ΔH°, ΔH⧧, ΔG°, ΔG⧧) have been determined for all stationary points along the step-growth mechanism of thiol–ene reactions between methyl mercaptan and a series of 12 alkenes: propene, methyl vinyl ether, methyl allyl ether, norbornene, acrylonitrile, methyl acrylate, butadiene, methyl(vinyl)silanediamine, methyl crotonate, dimethyl fumarate, styrene, and maleimide. Electronic structure calculations reveal the underlying factors that control activation barriers for propagation and chain-transfer processes of the step-growth mechanism. Results are further extended to predict rate constants for forward and reverse propagation and chain-transfer steps (kP, k–P, kCT, k–CT) and used to model overall reaction kinetics. A relationship between alkene structure and reactivity in thiol–ene reactions is derived from the results of kinetic modeling and can be directly related to the relative energetics of stationary points obtained from electronic structure calculations. The results predict the order of reactivity of alkenes and have broad implications for the use and applications of thiol–ene click chemistry.
Co-reporter:Merry K. Smith, Natalia E. Powers-Riggs and Brian H. Northrop
Chemical Communications 2013 - vol. 49(Issue 55) pp:NaN6169-6169
Publication Date(Web):2013/05/28
DOI:10.1039/C3CC42777C
The facile self-assembly of nanoscale boronate ester rectangles from linear bis-catechols and 1,4-benzene diboronic acid is described. Spectroscopic and computational analyses reveal the influence of extended π-conjugation on the rectangles' absorption and fluorescence properties. The rectangles represent a new class of discrete, organic soluble covalent organic polygons.

![Ethanol, 2,2'-[1,5-naphthalenediylbis(oxy-2,1-ethanediyloxy-2,1-ethanediyloxy-2,1-ethanediyloxy)]bis-](/data/chemimg/103300/168211-64-1.png)
![Ethanol, 2,2'-[1,5-naphthalenediylbis(oxy-2,1-ethanediyloxy-2,1-ethanediyloxy-2,1-ethanediyloxy)]bis-](/data/chemimg/103300/168211-64-1_b.png)








![6,9,12,15,18,29,32,35,38,41-Decaoxapentacyclo[40.4.0.05,46.019,24.023,28]hexatetraconta-1,3,5(46),19,21,23,25,27,42,44-decaene](/data/chemimg/103300/116059-04-2.png)
![6,9,12,15,18,29,32,35,38,41-Decaoxapentacyclo[40.4.0.05,46.019,24.023,28]hexatetraconta-1,3,5(46),19,21,23,25,27,42,44-decaene](/data/chemimg/103300/116059-04-2_b.png)
![Silane,[(4-ethynyl-1,2-phenylene)bis(oxy)]bis[(1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/109100/109032-33-9.png)
![Silane,[(4-ethynyl-1,2-phenylene)bis(oxy)]bis[(1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/109100/109032-33-9_b.png)
![Benzaldehyde, 3,4-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/99900/99815-16-4.png)
![Benzaldehyde, 3,4-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/99900/99815-16-4_b.png)
![Triphenylene, 2,3,6,7,10,11-hexakis[2-(2-methoxyethoxy)ethoxy]-](http://img.cochemist.com/ccimg/96500/96436-67-8.png)
![Triphenylene, 2,3,6,7,10,11-hexakis[2-(2-methoxyethoxy)ethoxy]-](http://img.cochemist.com/ccimg/96500/96436-67-8_b.png)