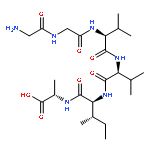
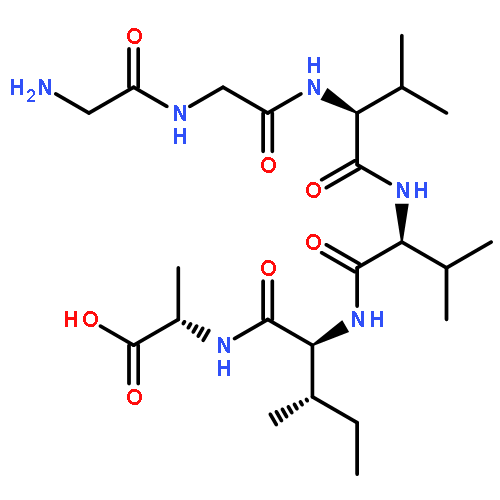
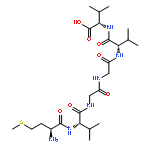
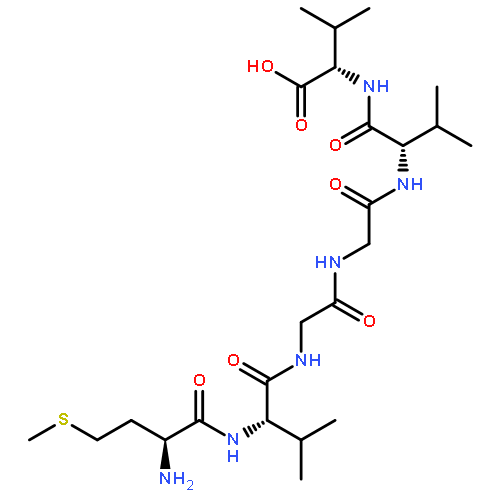
Co-reporter:Yuan Qiao;Mingzhen Zhang;Ya'nan Liang;Jie Zheng
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 1) pp:155-166
Publication Date(Web):2016/12/21
DOI:10.1039/C6CP07341G
The fibrillation and deposition of amyloid-β (Aβ) peptides in human brains are pathologically linked to Alzheimer's disease (AD). Development of different inhibitors (peptides, organic molecules, and nanoparticles) to prevent Aβ aggregation becomes a promising therapeutic strategy for AD treatment. We recently propose a “like-interacts-like” design principle to computationally design/screen and experimentally validate a new set of hexapeptide inhibitors with completely different sequences from the Aβ sequence. These hexapeptide inhibitors inhibit Aβ aggregation and reduce Aβ-induced cytotoxicity. However, inhibitory mechanisms of these hexapeptides and the underlying interactions between hexapeptides and Aβ remain unclear. Herein we apply multi-scale computational methods (quantum-chemical calculations, molecular docking and explicit-solvent molecular dynamic simulation) to explore the structure, dynamics, and interaction between 3 identified hexapeptides (CTLWWG, GTVWWG, and CTIYWG) and different Aβ-derived fragments and an Aβ17–42 pentamer. When interacting with 6 Aβ-derived fragments, 3 hexapeptide inhibitors show stronger interactions with two lysine-included fragments (16KLVFFA21 and 27NKGAII33) than other fragments, indicating different sequence-specific interactions with Aβ. When interacting with the Aβ17–42 pentamer, the 3 peptides show similar binding modes and interaction mechanisms by preferentially binding to the edge of the Aβ17–42 pentamer to potentially block the Aβ elongation pathway. This work provides structural-based binding information on further modification and optimization of these peptide inhibitors to experimentally enhance their inhibitory abilities against Aβ aggregation.
Co-reporter:Yu Qian, Ya’nan Liang, Wanqian Liu, Guizhao Liang
Journal of Molecular Graphics and Modelling 2017 Volume 71() pp:88-95
Publication Date(Web):January 2017
DOI:10.1016/j.jmgm.2016.11.003
•We compare twenty structural characterization scales applied as QSAM of AMPs.•Six methods have good characterization capabilities on the explored AMPs.•Hydrophobic, electrostatic and bulky properties are vital for activities of AMPs.•The FASGAI model exhibits a more robust predictive ability than other models.•The FASGAI model reasonably reveals the structure-activity relationship of AMPs.Informative numerical characterizations of amino acid residues are essential for quantitative sequence-activity modeling (QSAM). To date, a variety of structural characterization methods based on local amino acids have been proposed. However, limited detailed reports are available using same datasets and modeling methods to compare the ability to characterize structures of amino acids. Here, we evaluate the characterization capability of 20 descriptor sets on a set of antimicrobial peptides (AMPs) derived from Bac2A against P. aeruginosa. Results display the models by FASGAI, z-scales, VHSE, DPPS, HESH and ProtFP descriptors present qualified predictive capability. Moreover, the structural characterization of the studied AMPs should involve the hydrophobic, bulky and electronic properties of amino acids; besides, the secondary structural information should not be ignored. In parallel, the FASGAI-based model exhibits a more robust prediction than other models, and reasonably describe the structure-activity relationship of the studied dodecapeptides, which is in line with the reported experimental observations. This work provides references for methods of structural characterization as applied in QSAM of AMPs against P. aeruginosa.We evaluate the characterization capability of 20 descriptor sets on a set of antimicrobial peptides (AMPs) using variable selection by genetic algorithm and partial least squares modeling. This work provides references for methods of structural characterization as applied in QSAM of AMPs and other bioactive peptides.
Co-reporter:Chen Chen, Yonglan Liu, Jin Zhang, Mingzhen Zhang, Jie Zheng, Yong Teng, Guizhao Liang
Chemometrics and Intelligent Laboratory Systems 2015 Volume 145() pp:7-16
Publication Date(Web):15 July 2015
DOI:10.1016/j.chemolab.2015.04.009
•We build a quantitative sequence–aggregation relationship predictor.•We predict the self-assembled hexapeptides using the present model.•We investigate the characteristics of the self-assembled hexapeptides.•We examine the applications of the present model.It is essential to predict aggregation-forming sequences for elucidation of protein misfolding mechanisms and the design of effective antiamyloid inhibitors. In this work, we predict and characterize self-assembled hexapeptides by a quantitative sequence–aggregation relationship (QSAR) model, which involves characterization of factor analysis scale of generalized amino acid information (FASGAI) and modeling of supporting vector machine (SVM) with radial basis function kernel. The QSAR model achieves maximum accuracy of 78.33% and area under the receiver operating characteristic curve of 0.83 with leave one out cross-validation on 180 training hexapeptides. We determine “hotspots” and key factors that largely contribute to the self-assembly of these hexapeptides by analyzing their sequence–aggregation relationships. We also explore the applications of the present model, e.g., the first is to identify the aggregation-forming sequences within both β-amyloid peptide (Aβ42) and human islet amyloid polypeptide (hIAPP) using a 6-residue slide window, and acquire good agreement with previous experimental observations, the second is to perform in silico design of potential aggregation-forming hexapeptides which are validated by all-atom molecular dynamics simulation and density functional theory calculations, and the third is to predict the potential self-assembled tri-, tetra- and pentapeptides, in which hydrophobic amino acids such as isoleucine, leucine, valine, phenylalanine, and methionine occur at higher frequencies. The present QSAR model is helpful for (i) predicting self-assembled behaviors of peptides, (ii) scanning and identifying aggregation-forming sequences within proteins, (iii) understanding action mechanisms of peptide/protein aggregation, and (iv) designing potential self-assembled sequences applied as drug discovery and nano-materials.
Co-reporter:Yonglan Liu, Jin Zhang, Xiaohua Chen, Jie Zheng, Guixue Wang and Guizhao Liang
RSC Advances 2014 vol. 4(Issue 101) pp:58036-58046
Publication Date(Web):30 Oct 2014
DOI:10.1039/C4RA10195B
Exploring the adsorption characteristics of small molecules on carbon nanotubes (CNs) is important for rational design of CN-based materials for many applications. In this work, we construct a quantitative structure–activity relationship (QSAR) model to predict the adsorption of 25 simple benzene derivatives on CNs, and investigate the molecule–SWCN interactions by density functional theory (DFT) calculations, with the M062X functional at the 6-31G(d) basis set. The QSAR model exhibits a regression correlation coefficient (Rrm2) of 0.986 and a cross-validated coefficient (Qcv2) of 0.968. A total of more than 200 optimizations are carried out to determine preferential interaction modes, binding conformations, and underlying driving forces of the molecule–SWCN systems. We show the bridge configuration is the preferred molecule–SWCN interaction mode, which is mainly governed by π–π stacking; the molecules have experienced a significant electron rearrangement whereas the SWCNs have a weak one due to the molecule–SWCN mutual forces; and the substituents play dual effects on the adsorption in two ways, i.e., indirectly affecting π–π stacking by altering the electron density of the benzene ring and directly interacting with the nanotube surface. This work provides insights into noncovalent functionalization of CNs, and adsorption and desorption of simple organic molecules on CNs.
Co-reporter:Dandan Huang, Yonglan Liu, Bozhi Shi, Yueting Li, Guixue Wang, Guizhao Liang
Journal of Molecular Graphics and Modelling 2013 Volume 45() pp:65-83
Publication Date(Web):September 2013
DOI:10.1016/j.jmgm.2013.08.003
•We study 3D-QSAR of BACE-1 inhibitors and their binding mode.•We successfully design 30 new molecules with higher activity in theory.•We find the inhibitors closely interact with 10 key sites of BACE-1.The β-enzyme (BACE), which takes an active part in the processing of amyloid precursor protein, thereby leads to the production of amyloid-β (Aβ) in the brain, is a major therapeutic target against Alzheimer's disease. The present study is aimed at studying 3D-QSAR of BACE-1 inhibitors and their binding mode. We build a 3D-QSAR model involving 99 training BACE-1 inhibitors based on Topomer CoMFA, and 26 molecules are employed to validate the external predictive power of the model obtained. The multiple correlation coefficients of fitting modeling, leave one out cross validation, and external validation are 0.966, 0.767 and 0.784, respectively. Topomer search is used as a tool for virtual screening in lead-like compounds of ZINC databases (2012); as a result, we successfully design 30 new molecules with higher activity than that of all training and test inhibitors. Besides, Surflex-dock is employed to explore binding mode of the inhibitors studied when acting with BACE-1 enzyme. The result shows that the inhibitors closely interact with the key sites related to ASP93, THR133, GLN134, ASP289, GLY291, THR292, THR293, ASN294, ARG296 and SER386 of BACE-1.The present study is aimed at studying 3D-QSAR of BACE-1 inhibitors and their binding mode based on Topomer CoMFA and molecular docking. We successfully design 30 new molecules with higher activity than that of all training and test inhibitors using Topomer search. The result shows that the inhibitors closely interact with the key sites related to ASP93, THR133, GLN134, ASP289, GLY291, THR292, THR293, ASN294, ARG296 and SER386 of BACE-1.
Co-reporter:Wei Zhao;YuZhen Chen;Li Yang
Science China Chemistry 2011 Volume 54( Issue 7) pp:1064-1071
Publication Date(Web):2011 July
DOI:10.1007/s11426-011-4299-6
An integrated approach is proposed to predict the chromatographic retention time of oligonucleotides based on quantitative structure-retention relationships (QSRR) models. First, the primary base sequences of oligonucleotides are translated into vectors based on scores of generalized base properties (SGBP), involving physicochemical, quantum chemical, topological, spatial structural properties, etc.; thereafter, the sequence data are transformed into a uniform matrix by auto cross covariance (ACC). ACC accounts for the interactions between bases at a certain distance apart in an oligonucleotide sequence; hence, this method adequately takes the neighboring effect into account. Then, a genetic algorithm is used to select the variables related to chromatographic retention behavior of oligonucleotides. Finally, a support vector machine is used to develop QSRR models to predict chromatographic retention behavior. The whole dataset is divided into pairs of training sets and test sets with different proportions; as a result, it has been found that the QSRR models using more than 26 training samples have an appropriate external power, and can accurately represent the relationship between the features of sequences and structures, and the retention times. The results indicate that the SGBP-ACC approach is a useful structural representation method in QSRR of oligonucleotides due to its many advantages such as plentiful structural information, easy manipulation and high characterization competence. Moreover, the method can further be applied to predict chromatographic retention behavior of oligonucleotides.
Co-reporter:G. Liang;L. Yang;L. Kang;H. Mei;Z. Li
Amino Acids 2009 Volume 37( Issue 4) pp:583-591
Publication Date(Web):2009 October
DOI:10.1007/s00726-008-0177-8
On the basis of exploratory factor analysis, six multidimensional patterns of 516 amino acid attributes, namely, factor analysis scales of generalized amino acid information (FASGAI) involving hydrophobicity, alpha and turn propensities, bulky properties, compositional characteristics, local flexibility and electronic properties, are proposed to represent structures of 48 bitter-tasting dipeptides and 58 angiotensin-converting enzyme inhibitors. Characteristic parameters related to bioactivities of the peptides studied are selected by genetic algorithm, and quantitative structure–activity relationship (QSAR) models are constructed by partial least square (PLS). Our results by a leave-one-out cross validation are compared with the previously known structure representation method and are shown to give slightly superior or comparative performance. Further, two data sets are divided into training sets and test sets to validate the characterization repertoire of FASGAI. Performance of the PLS models developed by training samples by a leave-one-out cross validation and external validation for test samples are satisfying. These results demonstrate that FASGAI is an effective representation technique of peptide structures, and that FASGAI vectors have many preponderant characteristics such as straightforward physicochemical information, high characterization competence and easy manipulation. They can be further applied to investigate the relationship between structures and functions of various peptides, even proteins.
Co-reporter:Gui-zhao Liang, Xiu-yan Ma, Yuan-chao Li, Feng-lin Lv, Li Yang
Biosystems (July 2011) Volume 105(Issue 1) pp:101-106
Publication Date(Web):July 2011
DOI:10.1016/j.biosystems.2011.03.008
Co-reporter:Benguo Liu, Huizhi Xiao, Jiaqi Li, Sheng Geng, Hanjun Ma, Guizhao Liang
Food Chemistry (1 August 2017) Volume 228() pp:1-6
Publication Date(Web):1 August 2017
DOI:10.1016/j.foodchem.2017.01.126
Co-reporter:Yuan Qiao, Mingzhen Zhang, Ya'nan Liang, Jie Zheng and Guizhao Liang
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 1) pp:NaN166-166
Publication Date(Web):2016/11/23
DOI:10.1039/C6CP07341G
The fibrillation and deposition of amyloid-β (Aβ) peptides in human brains are pathologically linked to Alzheimer's disease (AD). Development of different inhibitors (peptides, organic molecules, and nanoparticles) to prevent Aβ aggregation becomes a promising therapeutic strategy for AD treatment. We recently propose a “like-interacts-like” design principle to computationally design/screen and experimentally validate a new set of hexapeptide inhibitors with completely different sequences from the Aβ sequence. These hexapeptide inhibitors inhibit Aβ aggregation and reduce Aβ-induced cytotoxicity. However, inhibitory mechanisms of these hexapeptides and the underlying interactions between hexapeptides and Aβ remain unclear. Herein we apply multi-scale computational methods (quantum-chemical calculations, molecular docking and explicit-solvent molecular dynamic simulation) to explore the structure, dynamics, and interaction between 3 identified hexapeptides (CTLWWG, GTVWWG, and CTIYWG) and different Aβ-derived fragments and an Aβ17–42 pentamer. When interacting with 6 Aβ-derived fragments, 3 hexapeptide inhibitors show stronger interactions with two lysine-included fragments (16KLVFFA21 and 27NKGAII33) than other fragments, indicating different sequence-specific interactions with Aβ. When interacting with the Aβ17–42 pentamer, the 3 peptides show similar binding modes and interaction mechanisms by preferentially binding to the edge of the Aβ17–42 pentamer to potentially block the Aβ elongation pathway. This work provides structural-based binding information on further modification and optimization of these peptide inhibitors to experimentally enhance their inhibitory abilities against Aβ aggregation.