Co-reporter:Ru-Yi Zhu, Luo-Yan Liu, and Jin-Quan Yu
Journal of the American Chemical Society September 13, 2017 Volume 139(Issue 36) pp:12394-12394
Publication Date(Web):August 27, 2017
DOI:10.1021/jacs.7b06851
The first example of palladium(II)-catalyzed β-C(sp3)–H iodination of a wide range of ketones using a commercially available aminooxyacetic acid auxiliary has been achieved. This L, X-type directing group overcomes the limitations of the transient directing group approach for C(sp3)–H functionalization of ketones. Practical advantages of this method include simple installation of the auxiliary without chromatography, exceptional tolerance of α-functional groups, as well as alkenes and alkynes, and rapid access to diverse sterically hindered quaternary centers.
Co-reporter:Kai Hong, Hojoon Park, and Jin-Quan Yu
ACS Catalysis October 6, 2017 Volume 7(Issue 10) pp:6938-6938
Publication Date(Web):September 13, 2017
DOI:10.1021/acscatal.7b02905
Palladium-catalyzed methylene β-C(sp3)–H arylation of aliphatic ketones using a transient directing group is developed. The use of α-benzyl β-alanine directing group that forms a six-membered chelation with palladium is crucial for promoting the methylene C(sp3)–H bond activation.Keywords: arylation; C−H activation; ketone; palladium; transient directing group; β-amino acid;
Co-reporter:Qing-Lan Pei, Guan-Da Che, Ru-Yi Zhu, Jian He, and Jin-Quan Yu
Organic Letters November 3, 2017 Volume 19(Issue 21) pp:5860-5860
Publication Date(Web):October 17, 2017
DOI:10.1021/acs.orglett.7b02841
A practical method for the removal of a versatile acidic amide auxiliary has been developed. Facile alcoholysis of the amide in the presence of KOAc is enabled by an epoxide, which mechanistically resembles the removal of the Myers’ auxiliary. The protocol has been applied to the removal of a variety of amide substrates and their C–H functionalization products with high efficiency and low cost, representing a step forward toward the development of a versatile directing group for C–H activation.
Co-reporter:Qian Shao, Jian He, Qing-Feng Wu, and Jin-Quan Yu
ACS Catalysis November 3, 2017 Volume 7(Issue 11) pp:7777-7777
Publication Date(Web):September 20, 2017
DOI:10.1021/acscatal.7b02721
Pd(II)-catalyzed γ-C(sp3)–H cross-coupling of 4-nitrobenzenesulfonyl (Ns)-protected amines is realized using both arylboron and alkylboron coupling partners. An acetyl-protected aminomethyl oxazoline (APAO) ligand is found to enable the C(sp3)–H arylation reaction, whereas mono-N-protected amino acid (MPAA) ligands promote the C(sp3)–H cross-coupling with various alkylboron reagents. Notably, the APAO-promoted C–H arylation reactions afford high diastereoselectivity (>20:1), providing a useful method for modifying chiral amines. The use of a common nosyl protecting group to direct C(sp3)–H activation significantly improves the practicality of this transformation, as demonstrated by the gram-scale stereoselective synthesis of γ-aryl- and γ-alkyl-α-amino acids.Keywords: amino acids; cross-coupling; C−H activation; palladium; synthetic methods;
Co-reporter:Jian He, Qian Shao, Qingfeng Wu, and Jin-Quan Yu
Journal of the American Chemical Society March 8, 2017 Volume 139(Issue 9) pp:3344-3344
Publication Date(Web):February 17, 2017
DOI:10.1021/jacs.6b13389
Pd(II)-catalyzed enantioselective borylation of C(sp3)–H bonds has been realized for the first time using chiral acetyl-protected aminomethyl oxazoline ligands. This reaction is compatible with carbocyclic amides containing α-tertiary as well as α-quaternary carbon centers. The chiral β-borylated amides are useful synthons for the synthesis of chiral β-hydroxylated, β-fluorinated, and β-arylated carboxylic acids.
Co-reporter:Yun-Fang Yang, Gang Chen, Xin Hong, Jin-Quan Yu, and K. N. Houk
Journal of the American Chemical Society June 28, 2017 Volume 139(Issue 25) pp:8514-8514
Publication Date(Web):June 3, 2017
DOI:10.1021/jacs.7b01801
The origin of the unique effectiveness of six-membered chelates on the β-methylene C(sp3)–H activation reactions by Pd(II) catalyst was explained with density functional theory. The Pd(II) catalysts that involve five-membered chelates are inactive in this transformation. Computational studies suggest that the C(sp3)–H bond activation is the rate-limiting step in both cases. The C(sp3)–H bond activation with a five-membered chelate is unfavorable by 7.7 kcal/mol compared to the corresponding six-membered chelate with Pd(II). Two factors cause the difference: (1) the dimeric Pd species with five-membered chelation square-planar structure is more stable than that with six-membered chelation by 2.0 kcal/mol; (2) steric repulsion between the ArF group of the substrate and the quinoline group of the acetyl-protected aminomethyl quinoline ligand destabilizes the five-membered chelate transition structure by 5.7 kcal/mol. The six-membered chelate of Pd(II) with an acetyl-protected aminoethyl quinoline ligand orients the ligand away from the ArF group of the substrate and alleviates the steric repulsion.
Co-reporter:Xing-Guo Zhang, Hui-Xiong Dai, Masayuki Wasa, and Jin-Quan Yu
Journal of the American Chemical Society July 25, 2012 Volume 134(Issue 29) pp:11948-11951
Publication Date(Web):July 11, 2012
DOI:10.1021/ja305259n
A Pd(II)-catalyzed trifluoromethylation of ortho C–H bonds with an array of N-arylbenzamides derived from benzoic acids is reported. N-Methylformamide has been identified as a crucial promoter of C–CF3 bond formation from the Pd center. X-ray characterization of the C–H insertion intermediate has revealed a rare coordination mode of acidic amides as directing groups and the origin of their capacity in directing C–H activation.
Co-reporter:Eun Jeong Yoo, Masayuki Wasa, and Jin-Quan Yu
Journal of the American Chemical Society December 15, 2010 Volume 132(Issue 49) pp:17378-17380
Publication Date(Web):November 17, 2010
DOI:10.1021/ja108754f
Pd(II)-catalyzed β-C(sp3)−H carbonylation of N-arylamides under CO (1 atm) has been achieved. Following amide-directed C(sp3)−H cleavage and insertion of CO into the resulting [Pd(II)−C(sp3)] bond, intramolecular C−N reductive elimination gave the corresponding succinimides, which could be readily converted to 1,4-dicarbonyl compounds. This method was found to be effective with substrates containing α-hydrogen atoms and could be applied to effect methylene C(sp3)−H carbonylation of cyclopropanes.
Co-reporter:Ru-Yi Zhu, Tyler G. Saint-Denis, Ying Shao, Jian He, Joshua D. Sieber, Chris H. Senanayake, and Jin-Quan Yu
Journal of the American Chemical Society April 26, 2017 Volume 139(Issue 16) pp:5724-5724
Publication Date(Web):April 10, 2017
DOI:10.1021/jacs.7b02196
We herein report the palladium(II)-catalyzed bromination and iodination of a variety of α-hydrogen-containing carboxylic acid and amino acid-derived amides. These reactions are exclusively enabled by quinoline-type ligands. The halogenated products obtained in this reaction are highly versatile and rapidly undergo further diversification. Further, we report the first example of a free carboxylic acid-directed Pd(II)-catalyzed C(sp3)–H bromination, enabled by quinoline ligands.
Co-reporter:Qing-Feng Wu;Peng-Xiang Shen;Jian He;Xiao-Bing Wang;Qian Shao;Ru-Yi Zhu;Forrest Zhang;Jennifer X. Qiao;Michael A. Poss;Claudio Mapelli
Science 2017 Volume 355(Issue 6324) pp:499-503
Publication Date(Web):03 Feb 2017
DOI:10.1126/science.aal5175
Expressed preferences among methyl groups
Targeting just one of the two equivalent branch ends in Y-shaped molecules is a particular challenge for catalysis. Enzymes manage to do it by grasping the whole molecule, octopus-like, but often enzymes cannot tolerate minor structural variations. Wu et al. produced an amide-directed palladium catalyst that, armed with oxazoline-derived chiral ligands, could reliably attack just one methyl member of isopropyl groups. The reaction successfully replaced C–H bonds with C–C bonds in a wide variety of aryl and vinyl coupling partners.
Science, this issue p. 499
Co-reporter:Ming Shang;Qian Shao;Shang-Zheng Sun;Yan-Qiao Chen;Hui Xu;Hui-Xiong Dai
Chemical Science (2010-Present) 2017 vol. 8(Issue 2) pp:1469-1473
Publication Date(Web):2017/01/30
DOI:10.1039/C6SC03383K
The use of a weakly coordinating monodentate directing group for copper mediated ortho-hydroxylation and amination reactions allows for the identification of an external oxazoline ligand as a promoter.
Co-reporter:Dr. Ming Shang;Ming-Ming Wang;Tyler G. Saint-Denis;Ming-Hong Li; Dr. Hui-Xiong Dai; Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2017 Volume 56(Issue 19) pp:5317-5321
Publication Date(Web):2017/05/02
DOI:10.1002/anie.201611287
AbstractOne long-standing issue in directed C−H functionalization is that either nitrogen or sulfur atoms present in heterocyclic substrates may bind preferentially to a transition-metal catalyst rather than to the desired directing group. This competitive binding has largely hindered the application of C−H functionalization in late-stage heterocycle drug discovery. Reported here is the use of an oxazoline-based directing group capable of overriding the poisoning effect of a wide range of heterocycle substrates. The potential use of this directing group in pharmaceutical drug discovery is illustrated by diversification of Telmisartan (an antagonist for the angiotensin II receptor) through copper-mediated C−H amination, hydroxylation, thiolation, arylation, and trifluoromethylation.
Co-reporter:Dr. Anh T. Tran; Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2017 Volume 56(Issue 35) pp:10530-10534
Publication Date(Web):2017/08/21
DOI:10.1002/anie.201704755
AbstractThe development of new and practical 3-pentoxythiocarbonyl auxiliaries for IrI-catalyzed C−H alkylation of azacycles is described. This method allows for the α-C−H alkylation of a variety of substituted pyrrolidines, piperidines, and tetrahydroisoquinolines through alkylation with alkenes. While the practicality of these simple carbamate-type auxiliaries is underscored by the ease of installation and removal, the method's utility is demonstrated in its ability to functionalize biologically relevant l-proline and l-trans-hydroxyproline, delivering unique 2,5-dialkylated amino acid analogues that are not accessible by other C−H functionalization methods.
Co-reporter:Dr. Anh T. Tran; Dr. Jin-Quan Yu
Angewandte Chemie 2017 Volume 129(Issue 35) pp:10666-10670
Publication Date(Web):2017/08/21
DOI:10.1002/ange.201704755
AbstractThe development of new and practical 3-pentoxythiocarbonyl auxiliaries for IrI-catalyzed C−H alkylation of azacycles is described. This method allows for the α-C−H alkylation of a variety of substituted pyrrolidines, piperidines, and tetrahydroisoquinolines through alkylation with alkenes. While the practicality of these simple carbamate-type auxiliaries is underscored by the ease of installation and removal, the method's utility is demonstrated in its ability to functionalize biologically relevant l-proline and l-trans-hydroxyproline, delivering unique 2,5-dialkylated amino acid analogues that are not accessible by other C−H functionalization methods.
Co-reporter:Yong-Qing Huang;Yue Zhao;Peng Wang;Taka-aki Okamura;Brian N. Laforteza;Yi Lu;Wei-Yin Sun
Dalton Transactions 2017 vol. 46(Issue 37) pp:12430-12433
Publication Date(Web):2017/09/25
DOI:10.1039/C7DT02883K
A new one-pot synthesis of C2-hydroxypropyl-substituted imidazolinium salts via the ring opening of tetrahydrofuran (THF) with N,N′-disubstituted diamines has been developed. Preliminary studies of the reaction mechanism suggest the CO2-promoted oxidative ring opening of THF followed by Hg(II)-mediated oxidation of an imidazolidine intermediate. These novel C2-substituted imidazolinium salts have shown to be active catalysts for the aza-Diels–Alder reactions.
Co-reporter:Gen-Cheng Li;Peng Wang;Marcus E. Farmer; Dr. Jin-Quan Yu
Angewandte Chemie 2017 Volume 129(Issue 24) pp:6978-6981
Publication Date(Web):2017/06/06
DOI:10.1002/ange.201702686
AbstractThe meta-C−H arylation of free phenylacetic acid was realized using 2-carbomethoxynorbornene (NBE-CO2Me) as a transient mediator. Both the modified norbornene and the mono-protected 3-amino-2-hydroxypyridine type ligand are crucial for this auxiliary-free meta-C−H arylation reaction. A series of phenylacetic acids, including mandelic acid and phenylglycine, react smoothly with various aryl iodides to provide the meta-arylated products in high yields.
Co-reporter: Dr. Guolin Cheng;Dr. Peng Wang; Dr. Jin-Quan Yu
Angewandte Chemie 2017 Volume 129(Issue 28) pp:8295-8298
Publication Date(Web):2017/07/03
DOI:10.1002/ange.201704411
AbstractPalladium(II)-catalyzed meta-C−H arylation and alkylation of benzylsulfonamide using 2-carbomethoxynorbornene (NBE-CO2Me) as a transient mediator are realized by using a newly developed electron-deficient directing group and isoquinoline as a ligand. This protocol features broad substrate scope and excellent functional-group tolerance. The meta-substituted benyzlsulfonamides can be readily transformed into sodium sulfonates, sulfonate esters, and sulfonamides, as well as styrenes by Julia-type olefination. The unique impact of the isoquinoline ligand underscores the importance of subtle matching between ligands and the directing groups.
Co-reporter:Dr. Peng Wang;Marcus E. Farmer; Dr. Jin-Quan Yu
Angewandte Chemie 2017 Volume 129(Issue 18) pp:5207-5211
Publication Date(Web):2017/04/24
DOI:10.1002/ange.201701803
AbstractMeta-C−H functionalization of benzylamines has been developed using a PdII/transient mediator strategy. Using 2-pyridone ligands and 2-carbomethoxynorbornene (NBE-CO2Me) as the mediator, arylation, amination, and chlorination of benzylamines are realized. This protocol features a broad substrate scope and is compatible with heterocylic coupling partners. Moreover, the loading of the Pd can be lowered to 2.5 mol % by using the optimal ligand.
Co-reporter:Tao Liu;Dr. Jennifer X. Qiao;Dr. Michael A. Poss; Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2017 Volume 56(Issue 36) pp:10924-10927
Publication Date(Web):2017/08/28
DOI:10.1002/anie.201706367
AbstractThe palladium(II)-catalyzed C(sp3)−H alkynylation of oligopeptides was developed with tetrabutylammonium acetate as a key additive. Through molecular design, the acetylene motif served as a linchpin to introduce a broad range of carbonyl-containing pharmacophores onto oligopeptides, thus providing a chemical tool for the synthesis and modification of novel oligopeptide–pharmacophore conjugates by C−H functionalization. Dipeptide conjugates with coprostanol and estradiol were synthesized by this method for potential application in targeted drug delivery to tumor cells with overexpressed nuclear hormone receptors.
Co-reporter:Huai-Wei Wang; Dr. Yi Lu;Bing Zhang;Dr. Jian He;Hua-Jin Xu;Yan-Shang Kang; Dr. Wei-Yin Sun; Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2017 Volume 56(Issue 26) pp:7449-7453
Publication Date(Web):2017/06/19
DOI:10.1002/anie.201703300
AbstractLigand development for rhodium(III)-catalyzed C−H activation reactions has largely been limited to cyclopentadienyl (Cp) based scaffolds. 2-Methylquinoline has now been identified as a feasible ligand that can coordinate to the metal center of Cp*RhCl to accelerate the cleavage of the C−H bond of N-pentafluorophenylbenzamides, providing a new structural lead for ligand design. The compatibility of this reaction with secondary free amines and anilines also overcomes the limitations of palladium(II)-catalyzed C−H amination reactions.
Co-reporter:Gen-Cheng Li;Peng Wang;Marcus E. Farmer; Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2017 Volume 56(Issue 24) pp:6874-6877
Publication Date(Web):2017/06/06
DOI:10.1002/anie.201702686
AbstractThe meta-C−H arylation of free phenylacetic acid was realized using 2-carbomethoxynorbornene (NBE-CO2Me) as a transient mediator. Both the modified norbornene and the mono-protected 3-amino-2-hydroxypyridine type ligand are crucial for this auxiliary-free meta-C−H arylation reaction. A series of phenylacetic acids, including mandelic acid and phenylglycine, react smoothly with various aryl iodides to provide the meta-arylated products in high yields.
Co-reporter: Dr. Guolin Cheng;Dr. Peng Wang; Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2017 Volume 56(Issue 28) pp:8183-8186
Publication Date(Web):2017/07/03
DOI:10.1002/anie.201704411
AbstractPalladium(II)-catalyzed meta-C−H arylation and alkylation of benzylsulfonamide using 2-carbomethoxynorbornene (NBE-CO2Me) as a transient mediator are realized by using a newly developed electron-deficient directing group and isoquinoline as a ligand. This protocol features broad substrate scope and excellent functional-group tolerance. The meta-substituted benyzlsulfonamides can be readily transformed into sodium sulfonates, sulfonate esters, and sulfonamides, as well as styrenes by Julia-type olefination. The unique impact of the isoquinoline ligand underscores the importance of subtle matching between ligands and the directing groups.
Co-reporter:Yongwei Wu, Yan-Qiao Chen, Tao Liu, Martin D. Eastgate, and Jin-Quan Yu
Journal of the American Chemical Society 2016 Volume 138(Issue 44) pp:14554-14557
Publication Date(Web):October 27, 2016
DOI:10.1021/jacs.6b09653
Pd(II)-catalyzed γ-C(sp3)–H arylation of primary amines is realized by using 2-hydroxynicotinaldehyde as a catalytic transient directing group. Importantly, the catalyst and the directing group loading can be lowered to 2% and 4% respectively, thus demonstrating high efficiency of this newly designed transient directing group. Heterocyclic aryl iodides are also compatible with this reaction. Furthermore, swift synthesis of 1,2,3,4-tetrahydronaphthyridine derivatives is accomplished using this reaction.
Co-reporter:Hang Shi, Peng Wang, Shin Suzuki, Marcus E. Farmer, and Jin-Quan Yu
Journal of the American Chemical Society 2016 Volume 138(Issue 45) pp:14876-14879
Publication Date(Web):November 2, 2016
DOI:10.1021/jacs.6b11055
Pd-catalyzed meta-C–H chlorination of anilines and phenols is developed using norbornene as the mediator. Heterocycles, including indole, thiophene, and indazole, are tolerated. The identification of a new pyridone-based ligand is crucial for the success of this meta-C–H chlorination reaction. Subsequent diverse transformations of the chlorinated products demonstrate the versatility of meta-C–H chlorination.
Co-reporter:Qiuping Ding, Shengqing Ye, Guolin Cheng, Peng Wang, Marcus E. Farmer, and Jin-Quan Yu
Journal of the American Chemical Society 2016 Volume 139(Issue 1) pp:417-425
Publication Date(Web):December 12, 2016
DOI:10.1021/jacs.6b11097
A Pd-catalyzed, meta-selective C–H arylation of nosyl-protected phenethylamines and benzylamines is disclosed using a combination of norbornene and pyridine-based ligands. Subjecting nosyl protected 2-aryl anilines to this protocol led to meta-C–H arylation at the remote aryl ring. A diverse range of aryl iodides are tolerated in this reaction, along with select heteroaryl iodides. Select aryl bromides bearing ortho-coordinating groups can also be utilized as effective coupling partners in this reaction. The use of pyridine ligands has allowed the palladium loading to be reduced to 2.5 mol %. Furthermore, a catalytic amount of 2-norbornene (20 mol %) to mediate this meta-C–H activation process is demonstrated for the first time. Utilization of a common protecting group as the directing group for meta-C–H activation of amines is an important feature of this reaction in terms of practical applications.
Co-reporter:Wei-Jun Kong; Yue-Jin Liu; Hui Xu; Yan-Qiao Chen; Hui-Xiong Dai
Journal of the American Chemical Society 2016 Volume 138(Issue 7) pp:2146-2149
Publication Date(Web):February 8, 2016
DOI:10.1021/jacs.5b13353
Pd-catalyzed α-olefinic C–H activation of simple α,β-unsaturated olefins has been developed. 4-imino-β-lactam derivatives were readily synthesized via activation of α-olefinic C–H bonds with excellent cis stereoselectivity. A wide range of heterocycles at the β-position are compatible with this reaction. The product of 4-imino-β-lactam derivatives can be readily converted to 2-aminoquinoline which exists extensively in pharmaceutical drugs and natural products.
Co-reporter:Peng Wang; Marcus E. Farmer; Xing Huo; Pankaj Jain; Peng-Xiang Shen; Mette Ishoey; James E. Bradner; Steven R. Wisniewski; Martin D. Eastgate
Journal of the American Chemical Society 2016 Volume 138(Issue 29) pp:9269-9276
Publication Date(Web):July 6, 2016
DOI:10.1021/jacs.6b04966
Here we report the development of a versatile 3-acetylamino-2-hydroxypyridine class of ligands that promote meta-C–H arylation of anilines, heterocyclic aromatic amines, phenols, and 2-benzyl heterocycles using norbornene as a transient mediator. More than 120 examples are presented, demonstrating this ligand scaffold enables a wide substrate and coupling partner scope. Meta-C–H arylation with heterocyclic aryl iodides as coupling partners is also realized for the first time using this ligand. The utility for this transformation for drug discovery is showcased by allowing the meta-C–H arylation of a lenalidomide derivative. The first steps toward a silver-free protocol for this reaction are also demonstrated.
Co-reporter:Heng Jiang; Jian He; Tao Liu
Journal of the American Chemical Society 2016 Volume 138(Issue 6) pp:2055-2059
Publication Date(Web):January 21, 2016
DOI:10.1021/jacs.5b13462
Pd(II)-catalyzed olefination of γ-C(sp3)–H bonds of triflyl (Tf) and 4-nitrobenzenesulfonyl (Ns) protected amines is achieved. Subsequent aza-Wacker oxidative cyclization or conjugate addition of the olefinated intermediates provides a variety of C-2 alkylated pyrrolidines. Three pyridine- and quinoline-based ligands are developed to match different classes of amine substrates, demonstrating a rare example of ligand-enabled C(sp3)–H olefination reactions. The use of Ns protecting group to direct C(sp3)–H activation of alkyl amines is also a significant step toward practical C–H functionalizations of alkyl amines.
Co-reporter:Xi-Hai Liu, Hojoon Park, Jun-Hao Hu, Yan Hu, Qun-Liang Zhang, Bao-Long Wang, Bing Sun, Kap-Sun Yeung, Fang-Lin Zhang, and Jin-Quan Yu
Journal of the American Chemical Society 2016 Volume 139(Issue 2) pp:888-896
Publication Date(Web):December 22, 2016
DOI:10.1021/jacs.6b11188
Pd-catalyzed C–H functionalizations promoted by transient directing groups remain largely limited to C–H arylation only. Herein, we report a diverse set of ortho-C(sp2)–H functionalizations of benzaldehyde substrates using the transient directing group strategy. Without installing any auxiliary directing group, Pd(II)-catalyzed C–H arylation, chlorination, bromination, and Ir(III)-catalyzed amidation, could be achieved on benzaldehyde substrates. The transient directing groups formed in situ via imine linkage can override other coordinating functional groups capable of directing C–H activation or catalyst poisoning, significantly expanding the scope for metal-catalyzed C–H functionalization of benzaldehydes. The utility of this approach is demonstrated through multiple applications, including late-stage diversification of a drug analogue.
Co-reporter:Peng Wang, Gen-Cheng Li, Pankaj Jain, Marcus E. Farmer, Jian He, Peng-Xiang Shen, and Jin-Quan Yu
Journal of the American Chemical Society 2016 Volume 138(Issue 42) pp:14092-14099
Publication Date(Web):October 7, 2016
DOI:10.1021/jacs.6b08942
Using a modified norbornene (methyl bicyclo[2.2.1]hept-2-ene-2-carboxylate) as a transient mediator, meta-C–H amination and meta-C–H alkynylation of aniline and phenol substrates have been developed for the first time. Both the identification of a monoprotected 3-amino-2-hydroxypyridine/pyridone-type ligand and the use of a modified norbornene as a mediator are crucial for the realization of these two unprecedented meta-C–H transformations. A variety of substrates are compatible with both meta-C–H amination and meta-C–H alkynylation. Amination and alkynylation of heterocyclic substrates including indole, indoline, and indazole afford the desired products in moderate to high yields.
Co-reporter:Kai-Jiong Xiao; Ling Chu; Gang Chen
Journal of the American Chemical Society 2016 Volume 138(Issue 24) pp:7796-7800
Publication Date(Web):June 1, 2016
DOI:10.1021/jacs.6b04660
A Pd(II)-catalyzed enantioselective C–H cross-coupling of benzylamines via kinetic resolution has been achieved using chiral mono-N-protected α-amino-O-methylhydroxamic acid (MPAHA) ligands. Both chiral benzylamines and ortho-arylated benzylamines are obtained in high enantiomeric purity. The use of a readily removable nosyl (Ns) protected amino group as the directing group is a crucial practical advantage. Moreover, the ortho-arylated benzylamine products could be further transformed into chiral 6-substituted 5,6-dihydrophenanthridines as important structural motifs in natural products and bioactive molecules.
Co-reporter:Li-Chen Lee, Jian He, Jin-Quan Yu, and Christopher W. Jones
ACS Catalysis 2016 Volume 6(Issue 8) pp:5245
Publication Date(Web):July 7, 2016
DOI:10.1021/acscatal.6b01774
The use of ligands to tune the reactivity and selectivity of transition-metal catalysts for C(sp3)–H bond activation is a current central challenge. One of us previously developed an uncommon example of a homogeneous catalyst that performs controlled C(sp3)–H arylation using pyridine derivatives as ligands, along with Pd [Science, 2014, 343, 1216–1220]. In this work, we report a functionalizable and tunable polymer support used in the immobilization of pyridine derivatives that yields a soluble, polymeric ligand platform facilitating C(sp3)–H activation reactions with good yields, selectivities differing from the homogeneous catalyst, and recovery of Pd. Unlike the homogeneous system, the supported catalysts in Pd-catalyzed C–H monoarylation reactions respond sensitively to the steric hindrance of the coupling partners.Keywords: arylation; C−H activation; immobilized catalyst; palladium; polymer-supported ligand
Co-reporter:Guo-Lin Gao, Wujiong Xia, Pankaj Jain, and Jin-Quan Yu
Organic Letters 2016 Volume 18(Issue 4) pp:744-747
Publication Date(Web):February 2, 2016
DOI:10.1021/acs.orglett.5b03712
Palladium catalyzed, nondirected C3-selective arylation of pyridines with arenes and heteroarenes in the presence of 1,10-phenanthroline as the ligand has been developed. The optimized conditions allow for a highly C3-selective arylation of pyridines, affording various 3,3′-bipyridines and 3-arylpyridines.
Co-reporter:Jin-Quan Yu;Fang-Lin Zhang;Tuan-Jie Li;Kai Hong;Hojoon Park
Science 2016 Volume 351(Issue 6270) pp:252-256
Publication Date(Web):15 Jan 2016
DOI:10.1126/science.aad7893
Amino acids can lend palladium a hand
Metals such as rhodium and palladium (Pd) are adept at activating otherwise inert carbon-hydrogen bonds toward useful reactivity. To get properly oriented, they often require some help from oxygen or nitrogen groups nearby. Appending and removing these directing groups, however, detracts from the efficiency of chemical synthesis. Zhang et al. show that amino acids can act as temporary directing groups in the Pd-catalyzed coupling of arenes with aldehydes or ketones. By reversibly binding to these latter substrates just long enough for the Pd catalysis to ensue, the amino acids eliminate the need for more laborious directing group manipulations.
Science, this issue p. 252
Co-reporter:Jian He;Heng Jiang;Ryosuke Takise;Ru-Yi Zhu;Dr. Gang Chen;Dr. Hui-Xiong Dai;Dr. T. G. Murali Dhar;Dr. Jun Shi;Dr. Hao Zhang;Dr. Peter T. W. Cheng;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2016 Volume 55( Issue 2) pp:785-789
Publication Date(Web):
DOI:10.1002/anie.201509996
Abstract
A quinoline-based ligand effectively promotes the palladium-catalyzed borylation of C(sp3)H bonds. Primary β-C(sp3)H bonds in carboxylic acid derivatives as well as secondary C(sp3)H bonds in a variety of carbocyclic rings, including cyclopropanes, cyclobutanes, cyclopentanes, cyclohexanes, and cycloheptanes, can thus be borylated. This directed borylation method complements existing iridium(I)- and rhodium(I)-catalyzed CH borylation reactions in terms of scope and operational conditions.
Co-reporter:Dr. Suhua Li;Ru-Yi Zhu;Dr. Kai-Jiong Xiao ;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2016 Volume 55( Issue 13) pp:4317-4321
Publication Date(Web):
DOI:10.1002/anie.201512020
Abstract
PdII-catalyzed arylation of γ-C(sp3)−H bonds of aliphatic acid-derived amides was developed by using quinoline-based ligands. Various γ-aryl-α-amino acids were prepared from natural amino acids using this method. The influence of ligand structure on reactivity was also systematically investigated.
Co-reporter:Jian He;Heng Jiang;Ryosuke Takise;Ru-Yi Zhu;Dr. Gang Chen;Dr. Hui-Xiong Dai;Dr. T. G. Murali Dhar;Dr. Jun Shi;Dr. Hao Zhang;Dr. Peter T. W. Cheng;Dr. Jin-Quan Yu
Angewandte Chemie 2016 Volume 128( Issue 2) pp:795-799
Publication Date(Web):
DOI:10.1002/ange.201509996
Abstract
A quinoline-based ligand effectively promotes the palladium-catalyzed borylation of C(sp3)H bonds. Primary β-C(sp3)H bonds in carboxylic acid derivatives as well as secondary C(sp3)H bonds in a variety of carbocyclic rings, including cyclopropanes, cyclobutanes, cyclopentanes, cyclohexanes, and cycloheptanes, can thus be borylated. This directed borylation method complements existing iridium(I)- and rhodium(I)-catalyzed CH borylation reactions in terms of scope and operational conditions.
Co-reporter:Dr. Kai-Jiong Xiao;Ling Chu ;Dr. Jin-Quan Yu
Angewandte Chemie 2016 Volume 128( Issue 8) pp:2906-2910
Publication Date(Web):
DOI:10.1002/ange.201510808
Abstract
Significant progress has been made in the past decade regarding the development of enantioselective C−H activation reactions by desymmetrization. However, the requirement for the presence of two chemically identical prochiral C−H bonds represents an inherent limitation in scope. Reported is the first example of kinetic resolution by a palladium(II)-catalyzed enantioselective C−H activation and C−C bond formation, thus significantly expanding the scope of enantioselective C−H activation reactions.
Co-reporter:Dr. Weibo Yang;Dr. Shengqing Ye;Yvonne Schmidt;Dean Stamos;Dr. Jin-Quan Yu
Chemistry - A European Journal 2016 Volume 22( Issue 21) pp:7059-7062
Publication Date(Web):
DOI:10.1002/chem.201600704
Abstract
A Pd-catalyzed/N-heterocycle-directed C(sp3)−H olefination has been developed. The monoprotected amino acid ligand (MPAA) is found to significantly promote Pd-catalyzed C(sp3)−H olefination for the first time. Cu(OAc)2 instead of Ag+ salts are used as the terminal oxidant. This reaction provides a useful method for the synthesis of alkylated pyrazoles.
Co-reporter:Dr. Kai-Jiong Xiao;Ling Chu ;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2016 Volume 55( Issue 8) pp:2856-2860
Publication Date(Web):
DOI:10.1002/anie.201510808
Abstract
Significant progress has been made in the past decade regarding the development of enantioselective C−H activation reactions by desymmetrization. However, the requirement for the presence of two chemically identical prochiral C−H bonds represents an inherent limitation in scope. Reported is the first example of kinetic resolution by a palladium(II)-catalyzed enantioselective C−H activation and C−C bond formation, thus significantly expanding the scope of enantioselective C−H activation reactions.
Co-reporter:Dr. Suhua Li;Ru-Yi Zhu;Dr. Kai-Jiong Xiao ;Dr. Jin-Quan Yu
Angewandte Chemie 2016 Volume 128( Issue 13) pp:4389-4393
Publication Date(Web):
DOI:10.1002/ange.201512020
Abstract
PdII-catalyzed arylation of γ-C(sp3)−H bonds of aliphatic acid-derived amides was developed by using quinoline-based ligands. Various γ-aryl-α-amino acids were prepared from natural amino acids using this method. The influence of ligand structure on reactivity was also systematically investigated.
Co-reporter:Peng-Xiang Shen; Xiao-Chen Wang; Peng Wang; Ru-Yi Zhu
Journal of the American Chemical Society 2015 Volume 137(Issue 36) pp:11574-11577
Publication Date(Web):August 27, 2015
DOI:10.1021/jacs.5b08914
2-Carbomethoxynorbornene is identified as a more effective transient mediator to promote a Pd(II)-catalyzed meta-C(sp2)–H alkylation of amides with various alkyl iodides as well as arylation with previously incompatible aryl iodides. The use of a tailor-made quinoline ligand is also crucial for this reaction to proceed.
Co-reporter:Tao Liu; Tian-Sheng Mei
Journal of the American Chemical Society 2015 Volume 137(Issue 18) pp:5871-5874
Publication Date(Web):April 27, 2015
DOI:10.1021/jacs.5b02065
Aliphatic amides are selectively functionalized at the γ- and δ-positions through directed radical 1,5 and 1,6 H-abstractions, respectively. The initially formed γ- or δ-lactams are intercepted by N-iodosuccinimide and trimethylsilyl azide, leading to double and triple C–H functionalizations at the γ-, δ-, and ε-positions. This new reactivity is exploited to convert alkyls into amino alcohols and allylic amines.
Co-reporter:Jian He; Ryosuke Takise; Haiyan Fu
Journal of the American Chemical Society 2015 Volume 137(Issue 14) pp:4618-4621
Publication Date(Web):March 30, 2015
DOI:10.1021/jacs.5b00890
Pd(II)-catalyzed cross-coupling of C(sp3)–H bonds with organosilicon coupling partners has been achieved for the first time. The use of a newly developed quinoline-based ligand is essential for the cross-coupling reactions to proceed.
Co-reporter:Ru-Yi Zhu; Keita Tanaka; Gen-Cheng Li; Jian He; Hai-Yan Fu; Su-Hua Li
Journal of the American Chemical Society 2015 Volume 137(Issue 22) pp:7067-7070
Publication Date(Web):May 22, 2015
DOI:10.1021/jacs.5b04088
A quinoline-based ligand was shown to promote palladium-catalyzed β-C(sp3)–H fluorination for the first time. A range of unnatural enantiopure fluorinated α-amino acids were obtained through sequential β-C(sp3)–H arylation and subsequent stereoselective fluorination from readily available l-alanine.
Co-reporter:Brandon E. Haines; Huiying Xu; Pritha Verma; Xiao-Chen Wang; Jin-Quan Yu;Djamaladdin G. Musaev
Journal of the American Chemical Society 2015 Volume 137(Issue 28) pp:9022-9031
Publication Date(Web):July 2, 2015
DOI:10.1021/jacs.5b03410
Transition metal-catalyzed C–H bond halogenation is an important alternative to the highly utilized directed-lithiation methods and increases the accessibility of the synthetically valuable aryl halide compounds. However, this approach often requires impractical reagents, such as IOAc, or strong co-oxidants. Therefore, the development of methodology utilizing inexpensive oxidants and catalyst containing earth-abundant transition metals under mild experimental conditions would represent a significant advance in the field. Success in this endeavor requires a full understanding of the mechanisms and reactivity governing principles of this process. Here, we report intimate mechanistic details of the Pd(II)-catalyzed C–H iodination with molecular I2 as the sole oxidant. Namely, we elucidate the impact of the: (a) Pd-directing group (DG) interaction, (b) nature of oxidant, and (c) nature of the functionalized C–H bond [C(sp2)–H vs C(sp3)–H] on the Pd(II)/Pd(IV) redox and Pd(II)/Pd(II) redox-neutral mechanisms of this reaction. We find that both monomeric and dimeric Pd(II) species may act as an active catalyst during the reaction, which preferentially proceeds via the Pd(II)/Pd(II) redox-neutral electrophilic cleavage (EC) pathway for all studied substrates with a functionalized C(sp2)–H bond. In general, a strong Pd–DG interaction increases the EC iodination barrier and reduces the I–I oxidative addition (OA) barrier. However, the increase in Pd–DG interaction alone is not enough to make the mechanistic switch from EC to OA: This occurs only upon changing to substrates with a functionalized C(sp3)–H bond. We also investigated the impact of the nature of the electrophile on the C(sp2)–H bond halogenation. We predicted molecular bromine (Br2) to be more effective electrophile for the C(sp2)–H halogenation than I2. Subsequent experiments on the stoichiometric C(sp2)–H bromination by Pd(OAc)2 and Br2 confirmed this prediction.The findings of this study advance our ability to design more efficient reactions with inexpensive oxidants under mild experimental conditions.
Co-reporter:Navid Dastbaravardeh; Tetsuya Toba; Marcus E. Farmer
Journal of the American Chemical Society 2015 Volume 137(Issue 31) pp:9877-9884
Publication Date(Web):July 10, 2015
DOI:10.1021/jacs.5b04324
Pd-catalyzed C–H functionalization of mandelic acid and α-phenylglycine is reported. We have developed different protocols for the arylation, iodination, acetoxylation, and olefination of these substrates based on two different (Pd(II)/Pd(IV) and Pd(II)/Pd(0)) catalytic cycles. Four crucial features of these protocols are advantageous for practical applications. First, the α-hydroxyl and amino groups are protected with simple protecting groups such as acetates (Ac, Piv) and carbamates (Boc, Fmoc), respectively. Second, these protocols do not involve installation and removal of a directing group. Third, monoselectivity is accomplished. Fourth, no epimerization occurs at the vulnerable α-chiral centers.
Co-reporter:Guolin Cheng; Tuan-Jie Li
Journal of the American Chemical Society 2015 Volume 137(Issue 34) pp:10950-10953
Publication Date(Web):August 14, 2015
DOI:10.1021/jacs.5b07507
Pd(II)-catalyzed ortho-alkylation of benzoic acids with both terminal and internal epoxides affords 3,4-dihydroisocoumarins in one step. The presence of potassium countercations is crucial for this reaction. Monoprotected amino acid ligands significantly promote this reaction, enabling the development of a practical C–H alkylation reaction using 0.5 mol % Pd catalyst. The inversion of stereochemistry in the C–H alkylation step is consistent with a redox-neutral SN2 nucleophilic ring-opening process as opposed to a Pd(II)/Pd(IV) pathway.
Co-reporter:Gang Li; Li Wan; Guofu Zhang; Dasheng Leow; Jillian Spangler
Journal of the American Chemical Society 2015 Volume 137(Issue 13) pp:4391-4397
Publication Date(Web):March 13, 2015
DOI:10.1021/ja5126897
Ortho-C(sp2)–H olefination and acetoxylation of broadly useful synthetic building blocks phenylacetyl Weinreb amides, esters, and ketones are developed without installing an additional directing group. The interplay between the distal weak coordination and the ligand-acceleration is crucial for these reactions to proceed under mild conditions. The tolerance of longer distance between the target C–H bonds and the directing functional groups also allows for the functionalizations of more distal C–H bonds in hydrocinnamoyl ketones, Weinreb amides, and biphenyl Weinreb amides. Mechanistically, the coordination of these carbonyl groups and the bisdentate amino acid ligand with Pd(II) centers provides further evidence for our early hypothesis that the carbonyl groups of the potassium carboxylate are responsible for the directed C–H activation of carboxylic acids.
Co-reporter:Gang Chen; Toshihiko Shigenari; Pankaj Jain; Zhipeng Zhang; Zhong Jin; Jian He; Suhua Li; Claudio Mapelli; Michael M. Miller; Michael A. Poss; Paul M. Scola; Kap-Sun Yeung
Journal of the American Chemical Society 2015 Volume 137(Issue 9) pp:3338-3351
Publication Date(Web):February 19, 2015
DOI:10.1021/ja512690x
Pd-catalyzed β-C–H functionalizations of carboxylic acid derivatives using an auxiliary as a directing group have been extensively explored in the past decade. In comparison to the most widely used auxiliaries in asymmetric synthesis, the simplicity and practicality of the auxiliaries developed for C–H activation remains to be improved. We previously developed a simple N-methoxyamide auxiliary to direct β-C–H activation, albeit this system was not compatible with carboxylic acids containing α-hydrogen atoms. Herein we report the development of a pyridine-type ligand that overcomes this limitation of the N-methoxyamide auxiliary, leading to a significant improvement of β-arylation of carboxylic acid derivatives, especially α-amino acids. The arylation using this practical auxiliary is applied to the gram-scale syntheses of unnatural amino acids, bioactive molecules, and chiral bis(oxazoline) ligands.
Co-reporter:Kelvin S. L. Chan; Hai-Yan Fu
Journal of the American Chemical Society 2015 Volume 137(Issue 5) pp:2042-2046
Publication Date(Web):January 12, 2015
DOI:10.1021/ja512529e
C–H arylation via a Pd(II)/Pd(IV) catalytic cycle has been one of the most extensively studied C–H activation reactions since the 1990s. Despite the rapid development of this reaction in the past two decades, an enantioselective version has not been reported to date. Herein, we report a Pd(II)-catalyzed highly enantioselective (up to 99.5% ee) arylation of cyclopropyl C–H bonds with aryl iodides using mono-N-protected amino acid (MPAA) ligands, providing a new route for the preparation of chiral cis-aryl-cyclopropylmethylamines. The enantiocontrol is also shown to override the diastereoselectivity of chiral substrates.
Co-reporter:Jillian E. Spangler; Yoshihisa Kobayashi; Pritha Verma; Dong-Hui Wang
Journal of the American Chemical Society 2015 Volume 137(Issue 37) pp:11876-11879
Publication Date(Web):August 31, 2015
DOI:10.1021/jacs.5b06740
Pd(II)-catalyzed α-C(sp3)–H arylation of pyrrolidines, piperidines, azepanes, and N-methylamines with arylboronic acids has been developed for the first time. This transformation is applicable to wide arrays of pyrrolidines and boronic acids, including heteroaromatic boronic acids. A diastereoselective one-pot heterodiarylation of pyrrolidines has also been achieved.
Co-reporter:Yi Lu, Huai-Wei Wang, Jillian E. Spangler, Kai Chen, Pei-Pei Cui, Yue Zhao, Wei-Yin Sun and Jin-Quan Yu
Chemical Science 2015 vol. 6(Issue 3) pp:1923-1927
Publication Date(Web):08 Jan 2015
DOI:10.1039/C4SC03350G
The oxidative olefination of a broad array of arenes and heteroarenes with a variety of activated and unactivated olefins has be achieved via a rhodium(III)-catalyzed C–H activation reaction. The use of an N-pentafluorophenyl benzamide directing group is crucial for achieving catalytic turnovers in the presence of air as the sole oxidant without using a co-oxidant.
Co-reporter:Tetsuya Toba, Yi Hu, Anh T. Tran, and Jin-Quan Yu
Organic Letters 2015 Volume 17(Issue 24) pp:5966-5969
Publication Date(Web):December 4, 2015
DOI:10.1021/acs.orglett.5b02900
The Pd(II)-catalyzed arylation of unactivated β-C(sp3)–H bonds in α-hydroxy aliphatic acid with a variety of aryl iodides was developed utilizing an amino acid auxiliary as a directing group. This protocol provides access to biologically active β-arylated-α-hydroxy acid derivatives.
Co-reporter:Hong-Li Wang, Ming Shang, Shang-Zheng Sun, Zeng-Le Zhou, Brian N. Laforteza, Hui-Xiong Dai, and Jin-Quan Yu
Organic Letters 2015 Volume 17(Issue 5) pp:1228-1231
Publication Date(Web):February 19, 2015
DOI:10.1021/acs.orglett.5b00193
A new Cu(II)-catalyzed oxidative coupling of arenes with malonates has been developed using an amide-oxazoline directing group. The reaction proceeds via C(sp2)–H activation and malonate coupling, followed by intramolecular oxidative N–C bond formation. A variety of arenes bearing different substituents are shown to be compatible with this reaction.
Co-reporter:Hui Xu, Ming Shang, Hui-Xiong Dai, and Jin-Quan Yu
Organic Letters 2015 Volume 17(Issue 15) pp:3830-3833
Publication Date(Web):July 23, 2015
DOI:10.1021/acs.orglett.5b01802
In a Pd-catalyzed double C–H activation reaction, a pyridine-type ligand is identified, for the first time, to enable a highly para-selective C–H arylation of monosubstituted arenes. Excellent para-selectivity is achieved with a variety of arenes containing alkyl, methoxyl, and halo substituents.
Co-reporter:Dr. Youqian Deng ;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2015 Volume 54( Issue 3) pp:888-891
Publication Date(Web):
DOI:10.1002/anie.201409860
Abstract
meta-CH olefination of phenylacetic acid derivatives has been achieved using a commercially available nitrile-containing template. The identification of N-formyl-protected glycine as the ligand (Formyl-Gly-OH) was crucial for the development of this reaction. Versatility of the template approach in accommodating macrocyclopalladation processes with different ring sizes is demonstrated.
Co-reporter:Dr. Dajian Zhu;Dr. Guoqiang Yang;Jian He;Ling Chu;Dr. Gang Chen;Dr. Wei Gong;Dr. Ke Chen;Dr. Martin D. Eastgate;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2015 Volume 54( Issue 8) pp:2497-2500
Publication Date(Web):
DOI:10.1002/anie.201408651
Abstract
2,4,6-Trimethoxypyridine is identified as an efficient ligand for promoting a Pd-catalyzed ortho-CH amination of both benzamides and triflyl-protected benzylamines. This finding provides guidance for the development of ligands that can improve or enable PdII-catalyzed CH activation reactions directed by weakly coordinating functional groups.
Co-reporter:Jian He;Toshihiko Shigenari ;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2015 Volume 54( Issue 22) pp:6545-6549
Publication Date(Web):
DOI:10.1002/anie.201502075
Abstract
An intermolecular C(sp3)H amination using a Pd0/PAr3 catalyst was developed. The reaction begins with oxidative addition of R2NOBz to a Pd0/PAr3 catalyst and subsequent cleavage of a C(sp3)H bond by the generated PdNR2 intermediate. The catalytic cycle proceeds without the need for external oxidants in a similar manner to the extensively studied palladium(0)-catalyzed CH arylation reactions. The electron-deficient triarylphosphine ligand is crucial for this C(sp3)H amination reaction to occur.
Co-reporter:Dr. Brian N. Laforteza;Kelvin S. L. Chan ;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2015 Volume 54( Issue 38) pp:11143-11146
Publication Date(Web):
DOI:10.1002/anie.201505204
Abstract
The commonly used para-nitrobenzenesulfonyl (nosyl) protecting group is employed to direct the CH activation of amines for the first time. An enantioselective ortho-CH cross-coupling between nosyl-protected diarylmethylamines and arylboronic acid pinacol esters has been achieved utilizing chiral mono-N-protected amino acid (MPAA) ligands as a promoter.
Co-reporter:Dr. Dajian Zhu;Dr. Guoqiang Yang;Jian He;Ling Chu;Dr. Gang Chen;Dr. Wei Gong;Dr. Ke Chen;Dr. Martin D. Eastgate;Dr. Jin-Quan Yu
Angewandte Chemie 2015 Volume 127( Issue 8) pp:2527-2530
Publication Date(Web):
DOI:10.1002/ange.201408651
Abstract
2,4,6-Trimethoxypyridine is identified as an efficient ligand for promoting a Pd-catalyzed ortho-CH amination of both benzamides and triflyl-protected benzylamines. This finding provides guidance for the development of ligands that can improve or enable PdII-catalyzed CH activation reactions directed by weakly coordinating functional groups.
Co-reporter:Dr. Brian N. Laforteza;Kelvin S. L. Chan ;Dr. Jin-Quan Yu
Angewandte Chemie 2015 Volume 127( Issue 38) pp:11295-11298
Publication Date(Web):
DOI:10.1002/ange.201505204
Abstract
The commonly used para-nitrobenzenesulfonyl (nosyl) protecting group is employed to direct the CH activation of amines for the first time. An enantioselective ortho-CH cross-coupling between nosyl-protected diarylmethylamines and arylboronic acid pinacol esters has been achieved utilizing chiral mono-N-protected amino acid (MPAA) ligands as a promoter.
Co-reporter:Jian He;Toshihiko Shigenari ;Dr. Jin-Quan Yu
Angewandte Chemie 2015 Volume 127( Issue 22) pp:6645-6649
Publication Date(Web):
DOI:10.1002/ange.201502075
Abstract
An intermolecular C(sp3)H amination using a Pd0/PAr3 catalyst was developed. The reaction begins with oxidative addition of R2NOBz to a Pd0/PAr3 catalyst and subsequent cleavage of a C(sp3)H bond by the generated PdNR2 intermediate. The catalytic cycle proceeds without the need for external oxidants in a similar manner to the extensively studied palladium(0)-catalyzed CH arylation reactions. The electron-deficient triarylphosphine ligand is crucial for this C(sp3)H amination reaction to occur.
Co-reporter:Dr. Youqian Deng ;Dr. Jin-Quan Yu
Angewandte Chemie 2015 Volume 127( Issue 3) pp:902-905
Publication Date(Web):
DOI:10.1002/ange.201409860
Abstract
meta-CH olefination of phenylacetic acid derivatives has been achieved using a commercially available nitrile-containing template. The identification of N-formyl-protected glycine as the ligand (Formyl-Gly-OH) was crucial for the development of this reaction. Versatility of the template approach in accommodating macrocyclopalladation processes with different ring sizes is demonstrated.
Co-reporter:Shang-Zheng Sun, Ming Shang, Hong-Li Wang, Hai-Xia Lin, Hui-Xiong Dai, and Jin-Quan Yu
The Journal of Organic Chemistry 2015 Volume 80(Issue 17) pp:8843-8848
Publication Date(Web):August 11, 2015
DOI:10.1021/acs.joc.5b01351
A Cu(II)-mediated ortho-C–H hydroxylation using a removable directing group has been developed. The reaction exhibits considerable functional group tolerance. The use of O2 as an oxidant is crucial for the reactivity. Water is also found to significantly improve this reaction.
Co-reporter:Ling Chu, Ming Shang, Keita Tanaka, Qinghao Chen, Natalya Pissarnitski, Eric Streckfuss, and Jin-Quan Yu
ACS Central Science 2015 Volume 1(Issue 7) pp:394
Publication Date(Web):October 16, 2015
DOI:10.1021/acscentsci.5b00312
The pyridyl group has been extensively employed to direct transition-metal-catalyzed C–H activation reactions in the past half-century. The typical cyclic transition states involved in these cyclometalation processes have only enabled the activation of ortho-C–H bonds. Here, we report that pyridine is adapted to direct meta-C–H activation of benzyl and phenyl ethyl alcohols through engineering the distance and geometry of a directing template. This template takes advantage of a stronger σ-coordinating pyridine to recruit Pd catalysts to the desired site for functionalization. The U-shaped structure accommodates the otherwise highly strained cyclophane-like transition state. This development illustrates the potential of achieving site selectivity in C–H activation via the recognition of distal and geometric relationship between existing functional groups and multiple C–H bonds in organic molecules.
Co-reporter:Kai-Jiong Xiao ; David W. Lin ; Motofumi Miura ; Ru-Yi Zhu ; Wei Gong ; Masayuki Wasa
Journal of the American Chemical Society 2014 Volume 136(Issue 22) pp:8138-8142
Publication Date(Web):May 9, 2014
DOI:10.1021/ja504196j
An enantioselective method for Pd(II)-catalyzed cross-coupling of methylene β-C(sp3)–H bonds in cyclobutanecarboxylic acid derivatives with arylboron reagents is described. High yields and enantioselectivities were achieved through the development of chiral mono-N-protected α-amino-O-methylhydroxamic acid (MPAHA) ligands, which form a chiral complex with the Pd(II) center. This reaction provides an alternative approach to the enantioselective synthesis of cyclobutanecarboxylates containing α-chiral quaternary stereocenters. This new class of chiral catalysts also show promises for enantioselective β-C(sp3)–H activation of acyclic amides.
Co-reporter:Ming Shang ; Hong-Li Wang ; Shang-Zheng Sun ; Hui-Xiong Dai
Journal of the American Chemical Society 2014 Volume 136(Issue 33) pp:11590-11593
Publication Date(Web):August 1, 2014
DOI:10.1021/ja507704b
Cu(II)-promoted ortho alkynylation of arenes and heteroarenes with terminal alkynes has been developed to prepare aryl alkynes. A variety of arenes and terminal alkynes bearing different substituents are compatible with this reaction, thus providing an alternative disconnection to Sonogashira coupling.
Co-reporter:Ru-Yi Zhu ; Jian He ; Xiao-Chen Wang
Journal of the American Chemical Society 2014 Volume 136(Issue 38) pp:13194-13197
Publication Date(Web):September 10, 2014
DOI:10.1021/ja508165a
9-Methylacridine was identified as a generally effective ligand to promote a Pd(II)-catalyzed C(sp3)–H and C(sp2)–H alkylation of simple amides with various alkyl iodides. This alkylation reaction was applied to the preparation of unnatural amino acids and geometrically controlled tri- and tetrasubstituted acrylic acids.
Co-reporter:Guoqiang Yang ; Petra Lindovska ; Dajian Zhu ; Justin Kim ; Peng Wang ; Ri-Yuan Tang ; Mohammad Movassaghi
Journal of the American Chemical Society 2014 Volume 136(Issue 30) pp:10807-10813
Publication Date(Web):July 9, 2014
DOI:10.1021/ja505737x
meta-C–H olefination, arylation, and acetoxylation of indolines have been developed using nitrile-containing templates. The combination of a monoprotected amino acid ligand and the nitrile template attached at the indolinyl nitrogen via a sulfonamide linkage is crucial for the meta-selective C–H functionalization of electron-rich indolines that are otherwise highly reactive toward electrophilic palladation at the para-positions. A wide range of synthetically important and advanced indoline analogues are selectively functionalized at the meta-positions.
Co-reporter:Suhua Li ; Gang Chen ; Chen-Guo Feng ; Wei Gong
Journal of the American Chemical Society 2014 Volume 136(Issue 14) pp:5267-5270
Publication Date(Web):March 26, 2014
DOI:10.1021/ja501689j
Monoselective γ-C–H olefination and carbonylation of aliphatic acids has been accomplished by using a combination of a quinoline-based ligand and a weakly coordinating amide directing group. The reaction provides a new route for constructing richly functionalized all-carbon quaternary carbon centers at the β-position of aliphatic acids.
Co-reporter:Ming Shang ; Shang-Zheng Sun ; Hui-Xiong Dai
Journal of the American Chemical Society 2014 Volume 136(Issue 9) pp:3354-3357
Publication Date(Web):February 17, 2014
DOI:10.1021/ja412880r
A Cu(OAc)2-mediated C–H amidation and amination of arenes and heteroarenes has been developed using a readily removable directing group. A wide range of sulfonamides, amides, and anilines function as amine donors in this reaction. Heterocycles present in both reactants are tolerated, making this a broadly applicable method for the synthesis of a family of inhibitors including 2-benzamidobenzoic acids and N-phenylaminobenzoates.
Co-reporter:Wei Gong ; Guofu Zhang ; Tao Liu ; Ramesh Giri
Journal of the American Chemical Society 2014 Volume 136(Issue 48) pp:16940-16946
Publication Date(Web):November 10, 2014
DOI:10.1021/ja510233h
Although the syntheses of novel and diverse peptides rely mainly on traditional coupling using unnatural amino acids, postsynthetic modification of peptides could provide a complementary method for the preparation of nonproteinogenic peptides. Site selectivity of postsynthetic modification of peptides is usually achieved by targeting reactive moieties, such as the thiol group of cysteine or the C-2 position of tryptophan. Herein, we report the development of site-selective functionalizations of inert C(sp3)–H bonds of N-terminal amino acids in di-, tri-, and tetrapeptides without installing a directing group. The native amino acid moiety within the peptide is used as a ligand to accelerate the C–H activation reaction. In the long run, this newly uncovered reactivity could provide guidance for developing site-selective C(sp3)–H activation toward postsynthetic modification of a broader range of peptides.
Co-reporter:Ming Shang, Shang-Zheng Sun, Hui-Xiong Dai, and Jin-Quan Yu
Organic Letters 2014 Volume 16(Issue 21) pp:5666-5669
Publication Date(Web):October 17, 2014
DOI:10.1021/ol5027377
Cu-catalyzed coupling of aryl C–H bonds with arylboron reagents was accomplished using a readily removable directing group, which provides a useful method for the synthesis of biaryl compounds. The distinct transmetalation step in this Cu-catalyzed C–H coupling with aryl borons provides unique evidence for the formation of an aryl cupperate intermediate.
Co-reporter:Jin-Quan Yu
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 7) pp:
Publication Date(Web):
DOI:10.1002/adsc.201400406
No abstract is available for this article.
Co-reporter:Marcus E. Farmer, Brian N. Laforteza, Jin-Quan Yu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 16) pp:4445-4452
Publication Date(Web):15 August 2014
DOI:10.1016/j.bmc.2014.05.031
In an idealistic setting, it can be imagined that if every CH bond on an organic molecule could be selectively functionalized, the fields of chemical synthesis and drug discovery would be forever revolutionized. With the purpose of investigating the practicality of this idealistic scenario, our group has endeavored to unlock the potential of nature’s CH bonds by developing palladium-catalyzed, site selective CH insertions that can be incorporated into both known and new catalytic cycles. To this end, we have developed a number of catalytic transformations that not only provide rapid diversification of simple starting materials and natural products through CH functionalization, but streamline the synthesis of a variety of natural products with biological activity and expand upon methods to access highly valuable enantiopure materials.
Co-reporter:Dr. Harit U. Vora;Anthony P. Silvestri;Casper J. Engelin ;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2014 Volume 53( Issue 10) pp:2683-2686
Publication Date(Web):
DOI:10.1002/anie.201310539
Abstract
A bimetallic RhII catalyst promoted the CH alkenylation of simple arenes at 1.0 equivalent without the use of a directing group. A phosphine ligand as well as cooperative reoxidation of RhII with Cu(TFA)2 and V2O5 proved essential in providing monoalkenylated products in good yields and selectivities, especially with di- and trisubstituted arenes.
Co-reporter:Dr. Sy Ma;Dr. Giorgio Villa;Peter S. Thuy-Boun;Anna Homs ;Dr. Jin-Quan Yu
Angewandte Chemie 2014 Volume 126( Issue 3) pp:753-756
Publication Date(Web):
DOI:10.1002/ange.201305388
Abstract
We disclose a protocol for the palladium-catalyzed ortho-selective CH deuteration of arenes. Phenylacetic acids and benzoic acids are suitable substrates for this reaction. This reaction offers a catalytic route to ortho-deuterated phenylacetic acids and benzoic acids and demonstrates the sharp difference in reactivity of palladacycle intermediates held together by weak and strong coordination.
Co-reporter:Dr. Youqian Deng;Dr. Wei Gong;Jian He ;Dr. Jin-Quan Yu
Angewandte Chemie 2014 Volume 126( Issue 26) pp:6810-6813
Publication Date(Web):
DOI:10.1002/ange.201403878
Abstract
Diverse 4-aryl-2-quinolinones are prepared from propionamides in one pot by ligand-promoted triple sequential CH activation reactions and a stereospecific Heck reaction. In these cascade reactions, three new CC bonds and one CN bond are formed to rapidly build molecular complexity from propionic acid.
Co-reporter:Dr. Sy Ma;Dr. Giorgio Villa;Peter S. Thuy-Boun;Anna Homs ;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2014 Volume 53( Issue 3) pp:734-737
Publication Date(Web):
DOI:10.1002/anie.201305388
Abstract
We disclose a protocol for the palladium-catalyzed ortho-selective CH deuteration of arenes. Phenylacetic acids and benzoic acids are suitable substrates for this reaction. This reaction offers a catalytic route to ortho-deuterated phenylacetic acids and benzoic acids and demonstrates the sharp difference in reactivity of palladacycle intermediates held together by weak and strong coordination.
Co-reporter:Dr. Harit U. Vora;Anthony P. Silvestri;Casper J. Engelin ;Dr. Jin-Quan Yu
Angewandte Chemie 2014 Volume 126( Issue 10) pp:2721-2724
Publication Date(Web):
DOI:10.1002/ange.201310539
Abstract
A bimetallic RhII catalyst promoted the CH alkenylation of simple arenes at 1.0 equivalent without the use of a directing group. A phosphine ligand as well as cooperative reoxidation of RhII with Cu(TFA)2 and V2O5 proved essential in providing monoalkenylated products in good yields and selectivities, especially with di- and trisubstituted arenes.
Co-reporter:Dr. Youqian Deng;Dr. Wei Gong;Jian He ;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2014 Volume 53( Issue 26) pp:6692-6695
Publication Date(Web):
DOI:10.1002/anie.201403878
Abstract
Diverse 4-aryl-2-quinolinones are prepared from propionamides in one pot by ligand-promoted triple sequential CH activation reactions and a stereospecific Heck reaction. In these cascade reactions, three new CC bonds and one CN bond are formed to rapidly build molecular complexity from propionic acid.
Co-reporter:Ming Shang;Shang-Zheng Sun;Dr. Hong-Li Wang;Dr. Brian N. Laforteza;Dr. Hui-Xiong Dai;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2014 Volume 53( Issue 39) pp:10439-10442
Publication Date(Web):
DOI:10.1002/anie.201404822
Abstract
The direct ortho-trifluoromethylation of arenes, including heteroarenes, with TMSCF3 has been accomplished by a copper(II)-promoted CH activation reaction which completes within 30 minutes. Mechanistic investigations are consistent with the involvement of CH activation, rather than a simple electrophilic aromatic substitution (SEAr), as the key step.
Co-reporter:Jian He;Suhua Li;Youqian Deng;Haiyan Fu;Brian N. Laforteza;Jillian E. Spangler;Anna Homs
Science 2014 Volume 343(Issue 6176) pp:1216-1220
Publication Date(Web):14 Mar 2014
DOI:10.1126/science.1249198
A Palladium 1–2 Punch
Methods to replace carbon-hydrogen bonds directly with carbon-carbon bonds offer enticing prospects for streamlining the synthesis of organic compounds. The trouble is that it is hard to select any particular C-H bond and to avoid making complex mixtures of products. He et al. (p. 1216) report that a pair of powerful pyrimidine ligands induces a palladium catalyst to add aryl groups selectively to amino acid derivatives. One ligand promotes addition of a single aryl group to the β-carbon center; the other appends a second, potentially different aryl group to the same carbon—all in the same flask.
Co-reporter:Ling Chu;Kai-Jiong Xiao
Science 2014 Volume 346(Issue 6208) pp:451-455
Publication Date(Web):24 Oct 2014
DOI:10.1126/science.1258538
Ensuring handedness when breaking C-H bonds
Many organic compounds are chiral: They manifest two distinct mirror-image variants, or enantiomers. Kinetic resolution can transform one enantiomer to a desired product while leaving its mirror image unmodified. Chu et al. applied this strategy to a reaction that replaces aryl carbon–hydrogen bonds with carbon-iodine bonds. They used a chiral palladium catalyst that reacts selectively with just one of two enantiomers of various benzylamine derivatives. In medicinal chemistry, such selective synthesis of individual enantiomers is essential for screening interactions with chiral biomolecules such as proteins.
Science, this issue p. 451
Co-reporter:Hengbin Wang ; Gang Li ; Keary M. Engle ; Jin-Quan Yu ;Huw M. L. Davies
Journal of the American Chemical Society 2013 Volume 135(Issue 18) pp:6774-6777
Publication Date(Web):April 20, 2013
DOI:10.1021/ja401731d
The enantioselective synthesis of 2,3-dihydrobenzofurans was achieved by using two sequential C–H functionalization reactions, a rhodium-catalyzed enantioselective intermolecular C–H insertion followed by a palladium-catalyzed C–H activation/C–O cyclization. Further diversification of the 2,3-dihydrobenzofuran structures was possible by a subsequent palladium-catalyzed intermolecular Heck-type sp2 C–H functionalization.
Co-reporter:Gui-Juan Cheng ; Yun-Fang Yang ; Peng Liu ; Ping Chen ; Tian-Yu Sun ; Gang Li ; Xinhao Zhang ; K. N. Houk ; Jin-Quan Yu ;Yun-Dong Wu
Journal of the American Chemical Society 2013 Volume 136(Issue 3) pp:894-897
Publication Date(Web):December 30, 2013
DOI:10.1021/ja411683n
A combined experimental/computational study on the amino acid ligand-assisted Pd-catalyzed C–H bond activation reveals a mechanism in which the amino acid acts as both a dianionic bidentate ligand and a proton acceptor. This new model explains the effects of amino acids on reactivity and selectivity and unveils the dual roles of amino acids: stabilizing monomeric Pd complexes and serving as the internal base for proton abstraction.
Co-reporter:Yun-Fang Yang ; Gui-Juan Cheng ; Peng Liu ; Dasheng Leow ; Tian-Yu Sun ; Ping Chen ; Xinhao Zhang ; Jin-Quan Yu ; Yun-Dong Wu ;K. N. Houk
Journal of the American Chemical Society 2013 Volume 136(Issue 1) pp:344-355
Publication Date(Web):December 8, 2013
DOI:10.1021/ja410485g
Density functional theory investigations have elucidated the mechanism and origins of meta-regioselectivity of Pd(II)-catalyzed C–H olefinations of toluene derivatives that employ a nitrile-containing template. The reaction proceeds through four major steps: C–H activation, alkene insertion, β-hydride elimination, and reductive elimination. The C–H activation step, which proceeds via a concerted metalation–deprotonation (CMD) pathway, is found to be the rate- and regioselectivity-determining step. For the crucial C–H activation, four possible active catalytic species—monomeric Pd(OAc)2, dimeric Pd2(OAc)4, heterodimeric PdAg(OAc)3, and trimeric Pd3(OAc)6—have been investigated. The computations indicated that the C–H activation with the nitrile-containing template occurs via a Pd–Ag heterodimeric transition state. The nitrile directing group coordinates with Ag while the Pd is placed adjacent to the meta-C–H bond in the transition state, leading to the observed high meta-selectivity. The Pd2(OAc)4 dimeric mechanism also leads to the meta-C–H activation product but with higher activation energies than the Pd–Ag heterodimeric mechanism. The Pd monomeric and trimeric mechanisms require much higher activation free energies and are predicted to give ortho products. Structural and distortion energy analysis of the transition states revealed significant effects of distortions of the template on mechanism and regioselectivity, which provided hints for further developments of new templates.
Co-reporter:Jian He ; Masayuki Wasa ; Kelvin S. L. Chan
Journal of the American Chemical Society 2013 Volume 135(Issue 9) pp:3387-3390
Publication Date(Web):February 13, 2013
DOI:10.1021/ja400648w
The alkynylation of β-C(sp3)–H bonds in aliphatic amides with alkynyl halides has been enabled using Pd(0)/N-heterocyclic carbene (NHC) and Pd(0)/phosphine (PR3) catalysts. This is the first example of utilizing [AlkynylPd(II)Ln] complexes to activate C(sp3)–H bonds.
Co-reporter:Hui-Xiong Dai ; Gang Li ; Xing-Guo Zhang ; Antonia F. Stepan
Journal of the American Chemical Society 2013 Volume 135(Issue 20) pp:7567-7571
Publication Date(Web):April 24, 2013
DOI:10.1021/ja400659s
A combination of weakly coordinating auxiliaries and ligand acceleration allows for the development of both ortho- and meta-selective C–H olefination of phenol derivatives. These reactions demonstrate the feasibility of directing C–H functionalizations when functional groups are distal to target C–H bonds. The meta-C–H functionalization of electron-rich phenol derivatives is unprecedented and orthogonal to previous electrophilic substitution of phenols in terms of regioselectivity. These methods are also applied to functionalize α-phenoxyacetic acids, a fibrate class of drug scaffolds.
Co-reporter:Chen-Guo Feng ; Mengchun Ye ; Kai-Jiong Xiao ; Suhua Li
Journal of the American Chemical Society 2013 Volume 135(Issue 25) pp:9322-9325
Publication Date(Web):June 11, 2013
DOI:10.1021/ja404526x
A Pd(II)-catalyzed C–H phosphorylation reaction has been developed using heterocycle-directed ortho-palladation. Both H-phosphonates and diaryl phosphine oxides are suitable coupling partners for this reaction.
Co-reporter:Xiao-Chen Wang ; Yi Hu ; Samuel Bonacorsi ; Yang Hong ; Richard Burrell
Journal of the American Chemical Society 2013 Volume 135(Issue 28) pp:10326-10329
Publication Date(Web):July 9, 2013
DOI:10.1021/ja4055492
Pd-catalyzed ortho-C–H iodination directed by a weakly coordinating amide auxiliary using I2 as the sole oxidant was developed. This reaction is compatible with a wide range of heterocycles including pyridines, imidazoles, oxazoles, thiazoles, isoxazoles, and pyrazoles.
Co-reporter:Travis M. Figg ; Masayuki Wasa ; Jin-Quan Yu ;Djamaladdin G. Musaev
Journal of the American Chemical Society 2013 Volume 135(Issue 38) pp:14206-14214
Publication Date(Web):September 4, 2013
DOI:10.1021/ja4053416
Mechanistic details pertaining to the Pd0/PCy3-catalyzed intermolecular arylation of a terminal β-C(sp3)–H bond aryl amide substrate (SM = EtCONH-Ar, where Ar = C6H5, C6F5 and CONH-Ar is a directing group (DG)) in the presence of CsF base were elucidated. Key mechanistic features of this reaction are (1) oxidative addition of the aryl halide PhI to Pd0/PCy3, (2) deprotonation of SM by CsF to form DG′ = [EtCON-Ar]Cs+ for subsequent coordination to intermediate I–PdII(PCy3)Ph (the substantially lower pKa of the EtCONHC6F5 in comparison to EtCONHC6H5 is instrumental for the presence of a larger population of the reactive deprotonated amides for Ar = C6F5), (3) “Cs2–I–F” cluster formation upon external (the second) CsF molecule approach to the active site of the I–PdII(PCy3)Ph(DG′) intermediate, (4) “Cs2–I–F cluster” assisted β-C(sp3)–H bond activation via a concerted metalation–deprotonation (CMD) mechanism, and (5) reprotonation of the amide directing group to facilitate the C(sp3)–Ph reductive elimination. The energy barriers, ΔG⧧ (ΔG⧧disp), associated with the “Cs2–I–F cluster” mediated β-C(sp3)–H bond activation transition state are 6.5 (8.7) and 10.2 (12.9) kcal/mol when DG = CONHC6H5, CONHC6F5, respectively. It was shown that (a) the PCy3 ligand only semidissociates upon β-C(sp3)–H bond cleavage and (b) the I-to-F substitution in I–[PdII](Ph)(PCy3)(DG′) is a facile process that makes the “direct-halide” assisted β-C(sp3)–H bond activation relatively less energy demanding and opens the possibility for a competing Ph–F bond formation reaction. It was shown that the “direct-I” assisted C–H bond activation TS, which associates with a relatively large energy barrier, is an H-atom insertion transition state into the Pd–I bond, while the “direct-F” assisted C–H bond activation TS, which occurs with a relatively low energy barrier (but still is much larger than that required for the “Cs2–I–F cluster” assisted pathway), is a direct proton abstraction transition state.
Co-reporter:Ling Chu ; Xiao-Chen Wang ; Curtis E. Moore ; Arnold L. Rheingold
Journal of the American Chemical Society 2013 Volume 135(Issue 44) pp:16344-16347
Publication Date(Web):October 23, 2013
DOI:10.1021/ja408864c
An enantioselective C–H iodination reaction using a mono-N-benzoyl-protected amino acid has been developed for the synthesis of chiral diarylmethylamines. The reaction uses iodine as the sole oxidant and proceeds at ambient temperature and under air.
Co-reporter:Peter S. Thuy-Boun ; Giorgio Villa ; Devin Dang ; Paul Richardson ; Shun Su
Journal of the American Chemical Society 2013 Volume 135(Issue 46) pp:17508-17513
Publication Date(Web):October 14, 2013
DOI:10.1021/ja409014v
A protocol for the Pd(II)-catalyzed ortho-C–H alkylation of phenylacetic and benzoic acids using alkylboron reagents is disclosed. Monoprotected amino acid ligands (MPAA) were found to significantly promote reactivity. Both potassium alkyltrifluoroborates and alkylboronic acids were compatible coupling partners. The possibility of a radical alkyl transfer to Pd(II) was also investigated.
Co-reporter:Li Wan ; Navid Dastbaravardeh ; Gang Li
Journal of the American Chemical Society 2013 Volume 135(Issue 48) pp:18056-18059
Publication Date(Web):November 17, 2013
DOI:10.1021/ja410760f
meta-C–H arylation and methylation of 3-phenylpropanoic acid and phenolic derivatives were developed using an easily removable nitrile template. The combination of a weakly coordinating U-shaped template and mono-protected amino acid ligand was crucial for the cross-coupling of C–H bonds with organoborons.
Co-reporter:Mengchun Ye, Andrew J. F. Edmunds, James A. Morris, David Sale, Yejia Zhang and Jin-Quan Yu
Chemical Science 2013 vol. 4(Issue 6) pp:2374-2379
Publication Date(Web):18 Mar 2013
DOI:10.1039/C3SC50184A
C3-arylated indazole and pyrazoles are privileged structural motifs in agrochemicals and pharmaceuticals. C-3 C–H arylation of (1H) indazole and pyrazole has been a significant challenge due to the poor reactivity of the C-3 position. Herein, we report a practical Pd(II)/Phen catalyst and conditions for the direct C-3 arylation of indazole and pyrazole with ArI or ArBr without using Ag additives as halide scavengers. The use of toluene, chlorobenzene, trifluoromethylbenzene and mesitylene as the solvent was found to be crucial for the selectivity and reactivity. We further demonstrate the robustness of this protocol through the first total synthesis of nigellidine hydrobromide as well as the expedient preparation of heterocycles structurally related to pesticides and drug molecules.
Co-reporter:Ming Shang, Shao-Hang Zeng, Shang-Zheng Sun, Hui-Xiong Dai, and Jin-Quan Yu
Organic Letters 2013 Volume 15(Issue 20) pp:5286-5289
Publication Date(Web):October 7, 2013
DOI:10.1021/ol402515s
The Ru(II)-catalyzed ortho-C–H amination directed by a weakly coordinating amide auxiliary with O-benzoyl hydroxylamines at room temperature has been achieved. This reaction is compatible with heterocycles including pyrazole, thiophene, benzothiophene, furan, benzofuran, and indole.
Co-reporter:Masanori Miura, Chen-Guo Feng, Sandy Ma, and Jin-Quan Yu
Organic Letters 2013 Volume 15(Issue 20) pp:5258-5261
Publication Date(Web):October 7, 2013
DOI:10.1021/ol402471y
The Pd(II)-catalyzed ortho-C–H trifluoromethylation of benzylamines has been achieved utilizing an electrophilic CF3 reagent. Additives, such as H2O and Ag2O, were found to be crucial for obtaining good yields. This protocol will be useful in medicinal chemistry for the preparation of ortho-trifluoromethyl-substituted benzylamines.
Co-reporter:Tian-Sheng Mei, Dasheng Leow, Han Xiao, Brian N. Laforteza, and Jin-Quan Yu
Organic Letters 2013 Volume 15(Issue 12) pp:3058-3061
Publication Date(Web):May 31, 2013
DOI:10.1021/ol401246u
The Pd(II)-catalyzed intramolecular C–H amination of 2-pyridinesulfonyl-protected phenethylamine derivatives has been achieved using PhI(OAc)2 as a bystanding oxidant, providing access to a variety of substituted indoline derivatives in good yields. The use of the 2-pyridinesulfonyl protecting group allows for facile deprotection following C–H functionalization.
Co-reporter:Dr. Gang Li;Dr. Dasheng Leow;Li Wan ;Dr. Jin-Quan Yu
Angewandte Chemie 2013 Volume 125( Issue 4) pp:1283-1285
Publication Date(Web):
DOI:10.1002/ange.201207770
Co-reporter:Keary M. Engle and Jin-Quan Yu
The Journal of Organic Chemistry 2013 Volume 78(Issue 18) pp:8927-8955
Publication Date(Web):April 8, 2013
DOI:10.1021/jo400159y
Homogeneous transition-metal-catalyzed reactions are indispensable to all facets of modern chemical synthesis. It is thus difficult to imagine that for much of the early 20th century, the reactivity and selectivity of all known homogeneous metal catalysts paled in comparison to their heterogeneous and biological counterparts. In the intervening decades, advances in ligand design bridged this divide, such that today some of the most demanding bond-forming events are mediated by ligand-supported homogeneous metal species. While ligand design has propelled many areas of homogeneous catalysis, in the field of Pd(II)-catalyzed C–H functionalization, suitable ligand scaffolds are lacking, which has hampered the development of broadly practical transformations based on C–H functionalization logic. In this Perspective, we offer an account of our research employing three ligand scaffolds, mono-N-protected amino acids, 2,6-disubstituted pyridines, and 2,2′-bipyridines, to address challenges posed by several synthetically versatile substrate classes. Drawing on this work, we discuss principles of ligand design, such as the need to match a ligand to a particular substrate class, and how ligand traits such as tunability and modularity can be advantageous in reaction discovery.
Co-reporter:Dr. Gang Li;Dr. Dasheng Leow;Li Wan ;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2013 Volume 52( Issue 4) pp:1245-1247
Publication Date(Web):
DOI:10.1002/anie.201207770
Co-reporter:Keary M. Engle, Tian-Sheng Mei, Masayuki Wasa, and Jin-Quan Yu
Accounts of Chemical Research 2012 Volume 45(Issue 6) pp:788
Publication Date(Web):December 14, 2011
DOI:10.1021/ar200185g
Reactions that convert carbon–hydrogen (C–H) bonds into carbon–carbon (C–C) or carbon–heteroatom (C–Y) bonds are attractive tools for organic chemists, potentially expediting the synthesis of target molecules through new disconnections in retrosynthetic analysis. Despite extensive inorganic and organometallic study of the insertion of homogeneous metal species into unactivated C–H bonds, practical applications of this technology in organic chemistry are still rare. Only in the past decade have metal-catalyzed C–H functionalization reactions become more widely utilized in organic synthesis.Research in the area of homogeneous transition metal–catalyzed C–H functionalization can be broadly grouped into two subfields. They reflect different approaches and goals and thus have different challenges and opportunities. One approach involves reactions of completely unfunctionalized aromatic and aliphatic hydrocarbons, which we refer to as “first functionalization”. Here the substrates are nonpolar and hydrophobic and thus interact very weakly with polar metal species. To overcome this weak affinity and drive metal-mediated C–H cleavage, chemists often use hydrocarbon substrates in large excess (for example, as solvent). Because highly reactive metal species are needed in first functionalization, controlling the chemoselectivity to avoid overfunctionalization is often difficult. Additionally, because both substrates and products are comparatively low-value chemicals, developing cost-effective catalysts with exceptionally high turnover numbers that are competitive with alternatives (including heterogeneous catalysts) is challenging. Although an exciting field, first functionalization is beyond the scope of this Account.The second subfield of C–H functionalization involves substrates containing one or more pre-existing functional groups, termed “further functionalization”. One advantage of this approach is that the existing functional group (or groups) can be used to chelate the metal catalyst and position it for selective C–H cleavage. Precoordination can overcome the paraffin nature of C–H bonds by increasing the effective concentration of the substrate so that it need not be used as solvent. From a synthetic perspective, it is desirable to use a functional group that is an intrinsic part of the substrate so that extra steps for installation and removal of an external directing group can be avoided. In this way, dramatic increases in molecular complexity can be accomplished in a single stroke through stereo- and site-selective introduction of a new functional group. Although reactivity is a major challenge (as with first functionalization), the philosophy in further functionalization differs; the major challenge is developing reactions that work with predictable selectivity in intricately functionalized contexts on commonly occurring structural motifs.In this Account, we focus on an emergent theme within the further functionalization literature: the use of commonly occurring functional groups to direct C–H cleavage through weak coordination. We discuss our motivation for studying Pd-catalyzed C–H functionalization assisted by weakly coordinating functional groups and chronicle our endeavors to bring reactions of this type to fruition. Through this approach, we have developed reactions with a diverse range of substrates and coupling partners, with the broad scope likely stemming from the high reactivity of the cyclopalladated intermediates, which are held together through weak interactions.
Co-reporter:Masayuki Wasa ; Kelvin S. L. Chan ; Xing-Guo Zhang ; Jian He ; Masanori Miura
Journal of the American Chemical Society 2012 Volume 134(Issue 45) pp:18570-18572
Publication Date(Web):November 1, 2012
DOI:10.1021/ja309325e
Pd(II) insertion into β-methylene C(sp3)–H bonds was enabled by a mutually repulsive and electron-rich quinoline ligand. Ligand tuning led to the development of a method that allows for installation of an aryl group on a range of acyclic and cyclic amides containing β-methylene C(sp3)–H bonds.
Co-reporter:Ramesh Giri ; Yu Lan ; Peng Liu ; K. N. Houk
Journal of the American Chemical Society 2012 Volume 134(Issue 34) pp:14118-14126
Publication Date(Web):July 25, 2012
DOI:10.1021/ja304643e
The origin of the high levels of reactivity and diastereoselectivity (>99:1 dr) observed in the oxazoline-directed, Pd(II)-catalyzed sp3 C–H bond iodination and acetoxylation reactions as reported in previous publications has been studied and explained on the basis of experimental and computational investigations. The characterization of a trinuclear chiral C–H insertion intermediate by X-ray paved the way for further investigations into C–H insertion step through the lens of stereochemistry. Computational investigations on reactivities and diastereoselectivities of C–H activation of t-Bu- and i-Pr-substituted oxazolines provided good agreement with the experimental results. Theoretical predictions with DFT calculations revealed that C–H activation occurs at the monomeric Pd center and that the most preferred transition state for C–H activation contains two sterically bulky t-Bu substituents in anti-positions due to steric repulsion and that this transition state leads to the major diastereomer, which is consistent with the structure of the newly characterized C–H insertion intermediate. The structural information about the transition state also suggests that a minimum dihedral angle between C–H bonds and Pd–OAc bonds is crucial for C–H bond cleavage. We have also utilized density functional theory (DFT) to calculate the energies of various potential intermediates and transition states with t-Bu- and i-Pr-substituted oxazolines and suggested a possible explanation for the substantial difference in reactivity between the t-Bu- and i-Pr-substituted oxazolines.
Co-reporter:Keary M. Engle ; Peter S. Thuy-Boun ; Michael Dang
Journal of the American Chemical Society 2011 Volume 133(Issue 45) pp:18183-18193
Publication Date(Web):September 12, 2011
DOI:10.1021/ja203978r
A ligand-accelerated Pd(II)-catalyzed C(sp2)–H/arylboron cross-coupling reaction of phenylacetic acid substrates is reported. Using Ac-Ile-OH as the ligand and Ag2CO3 as the oxidant, a fast, high-yielding, operationally simple, and functional group-tolerant protocol has been developed for the cross-coupling of phenylacetic acid substrates with aryltrifluoroborates. This ligand scaffold has also been shown to improve catalysis using 1 atm O2 as the sole reoxidant, which sheds light on the path forward in developing optimized ligands for aerobic C–H/arylboron cross-coupling.
Co-reporter:Mengchun Ye ; Guo-Lin Gao ; Andrew J. F. Edmunds ; P. A. Worthington ; James A. Morris
Journal of the American Chemical Society 2011 Volume 133(Issue 47) pp:19090-19093
Publication Date(Web):November 7, 2011
DOI:10.1021/ja209510q
The first example of Pd-catalyzed, C3-selective arylation of unprotected pyridines has been developed by employing a catalytic system consisting of Pd(OAc)2 and 1,10-phenanthroline. This protocol provides an expeditious route to an important class of 3-arylpyridines and 3-arylpiperidines frequently found in bioactive compounds. A brief synthesis of the drug molecule (±)-preclamol is also reported.
Co-reporter:Djamaladdin G. Musaev ; Alexey Kaledin ; Bing-Feng Shi
Journal of the American Chemical Society 2011 Volume 134(Issue 3) pp:1690-1698
Publication Date(Web):December 8, 2011
DOI:10.1021/ja208661v
Monoprotected chiral amino acids have recently been established as a class of ligand scaffolds for effecting Pd-catalyzed enantioselective C–H bond activation reactions. However, to elucidate the mechanistic details and controlling factors of these reactions, more comprehensive studies are needed. In this work we report computational investigations into the key mechanistic features of enantioselective C–H bond activation reactions catalyzed by a [chiral (mono-N-protected amino acid)–Pd(II)] complex. Structural analysis points to a C–H insertion intermediate in which the nitrogen atom of the ligand is bound as a neutral σ-donor. The formation of this C–H insertion intermediate could, in principle, proceed via a “direct C–H cleavage” or via “initial N–H bond cleavage followed by C–H cleavage”. The computational studies presented herein show that the pathway initiated by N–H bond cleavage is more kinetically favorable. It is shown that the first step of the reaction is the N–H bond cleavage by the coordinated acetate group (OAc). In the next stage, the weakly coordinated OAc– (the second acetate group) activates the ortho-C–H bond of the substrate and transfers the H-atom from the C-atom to the bound N-atom of the ligand. As a result, a new Pd–C bond is formed and the carbamate is converted from X-type to L-type ligand. The absolute configuration of the products that are predicted on the basis of the calculated energies of the transition states matches the experimental data. The calculated enantioselectivity is also comparable with the experimental result. On the basis of these data, the origin of the enantioselectivity can be largely attributed to steric repulsions in the transition states.
Co-reporter:Hui-Xiong Dai
Journal of the American Chemical Society 2011 Volume 134(Issue 1) pp:134-137
Publication Date(Web):December 9, 2011
DOI:10.1021/ja2097095
The development of a Pd-catalyzed oxidative ortho-C–H borylation with N-arylbenzamides is reported. A modified dibenzylideneacetone (dba) ligand, a weak base, and a strong oxidant are critical for obtaining good yields. The reaction is tolerant of electron-deficient and electron-rich benzamides derived from readily available benzoic acids. The borylated products can be converted to various synthons via diverse transformations.
Co-reporter:Xisheng Wang ; Dasheng Leow
Journal of the American Chemical Society 2011 Volume 133(Issue 35) pp:13864-13867
Publication Date(Web):August 11, 2011
DOI:10.1021/ja206572w
Pd-catalyzed highly para-selective C–H arylation of monosubstituted arenes (including toluene) is developed for the first time using an F+ reagent as a bystanding oxidant. This finding provides a new retrosynthetic disconnection for para-substituted biaryl synthesis via C–H/C–H cross-coupling.
Co-reporter:Dong-Hui Wang
Journal of the American Chemical Society 2011 Volume 133(Issue 15) pp:5767-5769
Publication Date(Web):March 28, 2011
DOI:10.1021/ja2010225
The total synthesis of (+)-lithospermic acid is reported, which exploits two successive C−H activation reactions as key steps. Rh-catalyzed carbene C−H insertion reaction utilizing Davies’s catalyst was used to forge dihydrobenzofuran core, and a late-stage intermolecular C−H olefination coupled the olefin unit with the dihydrobenzofuran core to construct the molecule in a highly convergent manner.
Co-reporter:Hui-Xiong Dai ; Antonia F. Stepan ; Mark S. Plummer ; Yang-Hui Zhang
Journal of the American Chemical Society 2011 Volume 133(Issue 18) pp:7222-7228
Publication Date(Web):April 13, 2011
DOI:10.1021/ja201708f
Modern drug discovery is contingent on identifying lead compounds and rapidly synthesizing analogues. The use of a common pharmacophore to direct multiple and divergent C–H functionalizations of lead compounds is a particularly attractive approach. Herein, we demonstrate the viability of late-stage diversification through the divergent C–H functionalization of sulfonamides, an important class of pharmacophores found in nearly 200 drugs currently on the market, including the non-steroidal anti-inflammatory blockbuster drug celecoxib. We developed a set of six categorically different sulfonamide C–H functionalization reactions (olefination, arylation, alkylation, halogenation, carboxylation, and carbonylation), each representing a distinct handle for further diversification to reach a large number of analogues. We then performed late-stage, site-selective diversification of a sulfonamide drug candidate containing multiple potentially reactive C–H bonds to synthesize directly novel celecoxib analogues as potential cyclooxygenase-II (COX-2)-specific inhibitors. Together with other recently developed practical directing groups, such as CONHOMe and CONHC6F5, sulfonamide directing groups demonstrate that the auxiliary approach established in asymmetric catalysis can be equally effective in developing broadly useful C–H activation reactions.
Co-reporter:Mengchun Ye ; Guo-Lin Gao
Journal of the American Chemical Society 2011 Volume 133(Issue 18) pp:6964-6967
Publication Date(Web):April 14, 2011
DOI:10.1021/ja2021075
Pd-catalyzed C-3 selective olefination of pyridines is developed for the first time using 1,10-phenanthroline as the ligand. This finding provides a novel disconnection for the synthesis of pyridine-containing alkaloids and drug molecules as well as a new approach for developing Pd-catalyzed C–H functionalizations of pyridines.
Co-reporter:Eun Jeong Yoo ; Sandy Ma ; Tian-Sheng Mei ; Kelvin S. L. Chan
Journal of the American Chemical Society 2011 Volume 133(Issue 20) pp:7652-7655
Publication Date(Web):April 26, 2011
DOI:10.1021/ja202563w
C–H amination of N-aryl benzamides with O-benzoyl hydroxylamines has been achieved with either Pd(II) or Pd(0) catalysts. Furthermore, we demonstrate that secondary amines can be directly used with benzoyl peroxide in a one-pot procedure that proceeds via the in situ generation of the appropriate O-benzoyl hydroxylamines. This catalytic reaction provides a new disconnection for the convergent synthesis of tertiary and secondary arylalkyl amines starting from benzoic acids.
Co-reporter:Yi Lu, Dasheng Leow, Xisheng Wang, Keary M. Engle and Jin-Quan Yu
Chemical Science 2011 vol. 2(Issue 5) pp:967-971
Publication Date(Web):18 Mar 2011
DOI:10.1039/C0SC00633E
A Pd(II)-catalyzed C–H carbonylation protocol of phenethyl alcohols has been developed using amino acid ligands to promote the reaction. This transformation provides an expedient route to 1-isochromanone motifs, which are common structural elements in natural products and other biologically active compounds. A concise synthesis of a histamine release inhibitor showcases the utility of this transformation.
Co-reporter:Kelvin S. L. Chan;Masayuki Wasa;Dr. Xisheng Wang ;Dr. Jin-Quan Yu
Angewandte Chemie 2011 Volume 123( Issue 39) pp:9247-9250
Publication Date(Web):
DOI:10.1002/ange.201102985
Co-reporter:Keary M. Engle;Tian-Sheng Mei;Dr. Xisheng Wang ; Jin-Quan Yu
Angewandte Chemie International Edition 2011 Volume 50( Issue 7) pp:1478-1491
Publication Date(Web):
DOI:10.1002/anie.201005142
Abstract
Reductive elimination from partially or completely oxidized metal centers is a vital step in a myriad of carbon–carbon and carbon–heteroatom bond-forming reactions. One strategy for promoting otherwise challenging reductive elimination reactions is to oxidize the metal center using a two-electron oxidant (that is, from M(n) to M(n+2)). However, many of the commonly used oxidants for this type of transformation contain oxygen, nitrogen, or halogen moieties that are subsequently capable of participating in reductive elimination, thus leading to a mixture of products. In this Minireview, we examine the use of bystanding F+ oxidants for addressing this widespread problem in organometallic chemistry and describe recent applications in PdII/PdIV and AuI/AuIII catalysis. We then briefly discuss a rare example in which one-electron oxidants have been shown to promote selective reductive elimination in palladium(II)-catalyzed CH functionalization, which we view as a promising future direction in the field.
Co-reporter:Kelvin S. L. Chan;Masayuki Wasa;Dr. Xisheng Wang ;Dr. Jin-Quan Yu
Angewandte Chemie International Edition 2011 Volume 50( Issue 39) pp:9081-9084
Publication Date(Web):
DOI:10.1002/anie.201102985
Co-reporter:Keary M. Engle;Tian-Sheng Mei;Dr. Xisheng Wang ; Jin-Quan Yu
Angewandte Chemie 2011 Volume 123( Issue 7) pp:1514-1528
Publication Date(Web):
DOI:10.1002/ange.201005142
Abstract
Die reduktive Eliminierung teils oder vollständig oxidierter Metallzentren ist ein wichtiger Schritt in einer Unzahl von C-C- und C-Heteroatom-Bindungsbildungen. Eine Möglichkeit, anspruchsvolle reduktive Eliminierungen zu erleichtern, besteht in der Oxidation der Metallzentren durch Zwei-Elektronen-Oxidationsmittel (also von Mn zu Mn+2). Viele der für diese Umwandlung typischen Oxidationsmittel enthalten allerdings Sauerstoff-, Stickstoff- oder Halogenatome — die sich anschließend an der reduktiven Eliminierung beteiligen könnten —, sodass ein Produktgemisch entstehen würde. In diesem Kurzaufsatz beschreiben wir die Untersuchungen zu einem neuartigen Lösungsansatz für dieses verbreitete Problem in der metallorganischen Chemie: die Verwendung von passiven F+-Oxidationsmitteln sowie die jüngsten Anwendungen in PdII/PdIV- und AuI/AuIII-Katalysen. Außerdem besprechen wir kurz ein seltenes Beispiel, in dem die Verwendung eines Ein-Elektronen-Oxidationsmittels zur Begünstigung einer selektiven reduktiven Eliminierung bei Palladium(II)-katalysierten C-H-Funktionalisierungen gezeigt werden konnte; dies sehen wir als eine vielversprechende Richtung für zukünftige Anwendungen auf diesem Gebiet an.
Co-reporter:Keary M. Engle ; Dong-Hui Wang
Journal of the American Chemical Society 2010 Volume 132(Issue 40) pp:14137-14151
Publication Date(Web):September 20, 2010
DOI:10.1021/ja105044s
Initial rate studies have revealed dramatic acceleration in aerobic Pd(II)-catalyzed C−H olefination reactions of phenylacetic acids when mono-N-protected amino acids are used as ligands. In light of these findings, systematic ligand tuning was undertaken, which has resulted in drastic improvements in substrate scope, reaction rate, and catalyst turnover. We present evidence from intermolecular competition studies and kinetic isotope effect experiments that implies that the observed rate increases are a result of acceleration in the C−H cleavage step. Furthermore, these studies suggest that the origin of this phenomenon is a change in the mechanism of C−H cleavage from electrophilic palladation to proton abstraction.
Co-reporter:Masayuki Wasa ; Keary M. Engle
Journal of the American Chemical Society 2010 Volume 132(Issue 11) pp:3680-3681
Publication Date(Web):February 26, 2010
DOI:10.1021/ja1010866
The first Pd(II)-catalyzed sp3 C−H olefination reaction has been developed using N-arylamide directing groups. Following olefination, the resulting intermediates were found to undergo rapid 1,4-addition to give the corresponding γ-lactams. Notably, this method was effective with substrates containing α-hydrogen atoms and could be applied to effect methylene C−H olefination of cyclopropane substrates.
Co-reporter:Xisheng Wang ; Larry Truesdale
Journal of the American Chemical Society 2010 Volume 132(Issue 11) pp:3648-3649
Publication Date(Web):February 25, 2010
DOI:10.1021/ja909522s
A Pd(II)-catalyzed C−H activation/trifluoromethylation of arenes with an electrophilic trifluoromethylation reagent using diverse heterocycle directing groups is reported. The presence of trifluoroacetic acid is crucial for this catalytic reaction.
Co-reporter:Xisheng Wang ; Yi Lu ; Hui-Xiong Dai
Journal of the American Chemical Society 2010 Volume 132(Issue 35) pp:12203-12205
Publication Date(Web):August 17, 2010
DOI:10.1021/ja105366u
A Pd(II)-catalyzed C−H activation/C−O cyclization reaction directed by a proximate hydroxyl group has been developed. This reaction provides a new method for constructing dihydrobenzofurans, including spirocyclic analogues, a process that is potentially applicable to natural product synthesis.
Co-reporter:Chris J. Vickers, Tian-Sheng Mei and Jin-Quan Yu
Organic Letters 2010 Volume 12(Issue 11) pp:2511-2513
Publication Date(Web):May 6, 2010
DOI:10.1021/ol1007108
Pd(II)-catalyzed ortho C−H acetoxylation of triflate protected phenethyl- and phenpropylamines has been achieved with tert-butyl peroxyacetate as the stoichiometric oxidant and either DMF or CH3CN as the promoter. The reaction was found to tolerate a large variety of functional groups and could be combined with subsequent intramolecular amination to afford functionalized indoline derivatives.
Co-reporter:Tian-Sheng Mei, Dong-Hui Wang and Jin-Quan Yu
Organic Letters 2010 Volume 12(Issue 14) pp:3140-3143
Publication Date(Web):June 15, 2010
DOI:10.1021/ol1010483
Pd(II)-catalyzed ortho-C−H iodination reactions of phenylacetic acid substrates have been achieved using recyclable PdI2 as the precatalyst. This class of substrates is incompatible with the classic amide formation/ortho-lithiation/iodination sequence. The power of this new technology is demonstrated by facile drug functionalization and drastically shortened syntheses of the drugs diclofenac and lumiracoxib.
Co-reporter:Masayuki Wasa;Keary M. Engle
Israel Journal of Chemistry 2010 Volume 50( Issue 5-6) pp:605-616
Publication Date(Web):
DOI:10.1002/ijch.201000038
Abstract
Palladium-catalyzed C–H activation/C–C bond-forming reactions have emerged as a promising class of synthetic tools in organic chemistry. Among the many different means of forging C–C bonds using Pd-mediated C–H activation, a new horizon in this field is Pd(II)-catalyzed cross-coupling of C–H bonds with organometallic reagents via a Pd(II)/Pd(0) catalytic cycle. While this type of reaction has proven to be effective for the selective functionalization of aryl C(sp2)–H bonds, the focus of this review is on Pd(II)-catalyzed C(sp3)–H activation/C–C cross-coupling, a topic of particular importance because reactions of this type enable fundamentally new methods for bond construction. Since our laboratory’s initial report on cross-coupling of C–H bonds in 2006, this area has expanded rapidly, and the unique ability of Pd(II) catalysts to cleave and functionalize alkyl C(sp3)–H bonds has been exploited to develop protocols for forming an array of C(sp3)–C(sp2) and C(sp3)–C(sp3) bonds. Furthermore, enantioselective C(sp3)–H activation/C–C cross-coupling has been achieved through the use of chiral amino acid-derived ligands, offering a novel technique for producing enantioenriched molecules. Although this nascent field remains at an early stage of development, further investigations hold the potential to revolutionalize the way in which chiral molecules are synthesized in industrial and academic laboratories.
Co-reporter:Masayuki Wasa;Keary M. Engle
Israel Journal of Chemistry 2010 Volume 50( Issue 5-6) pp:
Publication Date(Web):
DOI:10.1002/ijch.201090011
Co-reporter:Dong-Hui Wang;Keary M. Engle;Bing-Feng Shi
Science 2010 Volume 327(Issue 5963) pp:315-319
Publication Date(Web):15 Jan 2010
DOI:10.1126/science.1182512
Heck of an Alternative
The Mizoroki-Heck reaction is widely used in organic synthesis to link together unsaturated carbon fragments such as olefins and arenes. However, one of its drawbacks is the need to append a reactive group such as a halogen to one of the reagents beforehand. Wang et al. (p. 315, published online 26 November) present an alternative palladium-catalyzed reaction that links olefins directly to aryl acids. Oxygen added to the reaction medium concurrently oxidizes the aryl C-H bond at the linkage site, eliminating the need for prior halogenation. Introducing amino acid–derived ligands tunes the aryl site at which the reaction takes place, and efficient reactivity can be achieved across a diverse range of substrates.
Co-reporter:Masayuki Wasa;BradyT. Worrell Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 7) pp:1275-1277
Publication Date(Web):
DOI:10.1002/anie.200906104
Co-reporter:KearyM. Engle;Dong-Hui Wang
Angewandte Chemie International Edition 2010 Volume 49( Issue 35) pp:6169-6173
Publication Date(Web):
DOI:10.1002/anie.201002077
Co-reporter:Masayuki Wasa;BradyT. Worrell Dr.
Angewandte Chemie 2010 Volume 122( Issue 7) pp:1297-1299
Publication Date(Web):
DOI:10.1002/ange.200906104
Co-reporter:Masayuki Wasa, Jin-Quan Yu
Tetrahedron 2010 66(26) pp: 4811-4815
Publication Date(Web):
DOI:10.1016/j.tet.2010.03.111
Co-reporter:KearyM. Engle;Dong-Hui Wang
Angewandte Chemie 2010 Volume 122( Issue 35) pp:6305-6309
Publication Date(Web):
DOI:10.1002/ange.201002077
Co-reporter:Ramesh Giri, Bing-Feng Shi, Keary M. Engle, Nathan Maugel and Jin-Quan Yu
Chemical Society Reviews 2009 vol. 38(Issue 11) pp:3242-3272
Publication Date(Web):2009/10/01
DOI:10.1039/B816707A
This critical review discusses historical and contemporary research in the field of transition metal-catalyzed carbon–hydrogen (C–H) bond activation through the lens of stereoselectivity. Research concerning both diastereoselectivity and enantioselectivity in C–H activation processes is examined, and the application of concepts in this area for the development of novel carbon–carbon and carbon–heteroatom bond-forming reactions is described. Throughout this review, an emphasis is placed on reactions that are (or may soon become) relevant in the realm of organic synthesis (221 references).
Co-reporter:Bing-Feng Shi ; Yang-Hui Zhang ; Jonathan K. Lam ; Dong-Hui Wang
Journal of the American Chemical Society 2009 Volume 132(Issue 2) pp:460-461
Publication Date(Web):December 17, 2009
DOI:10.1021/ja909571z
Pd(II)-catalyzed enantioselective C−H olefination of diphenylacetic acid substrates has been achieved through the use of monoprotected chiral amino acid ligands. The absolute configuration of the resulting olefinated products is consistent with that of a proposed C−H insertion intermediate.
Co-reporter:Ramesh Giri ; Jonathan K. Lam
Journal of the American Chemical Society 2009 Volume 132(Issue 2) pp:686-693
Publication Date(Web):December 15, 2009
DOI:10.1021/ja9077705
A Pd(II)-catalyzed reaction protocol for the carboxylation of ortho-C−H bonds in anilides to form N-acyl anthranilic acids has been developed. This reaction procedure provides a novel and efficient strategy for the rapid assembly of biologically and pharmaceutically significant molecules, such as benzoxazinones and quinazolinones, from simple anilides without installing and removing an external directing group. The reaction conditions are also amenable to the carboxylation of N-phenyl pyrrolidinones. A monomeric palladacycle containing p-toluenesulfonate as an anionic ligand has been characterized by X-ray crystallography, and the crucial role of p-toluenesulfonic acid in the activation of C−H bonds in the presence of carbon monoxide is discussed. Identification of two key intermediates, a mixed anhydride and benzoxazinone formed by reductive elimination from organometallic Ar(CO)Pd(II)−OTs species, provides mechanistic evidence for a dual-reaction pathway.
Co-reporter:Hai-Chen Wu ; Shafida Abd Hamid ; Jin-Quan Yu ;Jonathan B. Spencer
Journal of the American Chemical Society 2009 Volume 131(Issue 28) pp:9604-9605
Publication Date(Web):June 25, 2009
DOI:10.1021/ja903089f
Reducing the electron density of ligands switches the regioselectivity of Rh(I)-catalyzed hydrometalation. A reversal of the sense of chiral induction was also observed when chiral ligands are electronically tuned in the same manner. The combined data provide an alternative rationale for the electronic effects often observed in asymmetric hydrogenation.
Co-reporter:Xisheng Wang ; Tian-Sheng Mei
Journal of the American Chemical Society 2009 Volume 131(Issue 22) pp:7520-7521
Publication Date(Web):May 12, 2009
DOI:10.1021/ja901352k
Pd(OTf)2·2H2O-catalyzed ortho-fluorination of triflamide-protected benzylamines is reported. The use of N-fluoro-2,4,6-trimethylpyridinium triflate as the F+ source and NMP as a promoter is crucial for this reaction. The conversion of triflamide into a wide range of synthetically useful functional groups makes this fluorination protocol broadly applicable in medicinal chemistry and synthesis.
Co-reporter:Yang-Hui Zhang
Journal of the American Chemical Society 2009 Volume 131(Issue 41) pp:14654-14655
Publication Date(Web):September 29, 2009
DOI:10.1021/ja907198n
Pd(II)-catalyzed ortho-hydroxylation of variously substituted benzoic acids under 1 atm of O2 or air is achieved under nonacidic conditions. Extensive labeling studies support a direct oxygenation of aryl C−H bonds with molecular oxygen.
Co-reporter:Tian-Sheng Mei ; Xisheng Wang
Journal of the American Chemical Society 2009 Volume 131(Issue 31) pp:10806-10807
Publication Date(Web):July 16, 2009
DOI:10.1021/ja904709b
Pd(II)-catalyzed intramolecular amination of arenes is developed using either a one- or two-electron oxidant. The reaction protocol tolerates a wide range of deactivating groups including acetyl, cyano, and nitro groups. This catalytic reaction allows expedient syntheses of broadly useful substituted indolines or indoles.
Co-reporter:Yang-Hui Zhang Dr.;Bing-Feng Shi Dr.
Angewandte Chemie 2009 Volume 121( Issue 33) pp:6213-6216
Publication Date(Web):
DOI:10.1002/ange.200902262
Co-reporter:Xiao Chen Dr.;KearyM. Engle;Dong-Hui Wang Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 28) pp:5094-5115
Publication Date(Web):
DOI:10.1002/anie.200806273
Co-reporter:Xiao Chen, Graham Dobereiner, Xue-Shi Hao, Ramesh Giri, Nathan Maugel, Jin-Quan Yu
Tetrahedron 2009 65(16) pp: 3085-3089
Publication Date(Web):
DOI:10.1016/j.tet.2008.10.053
Co-reporter:Yang-Hui Zhang Dr.;Bing-Feng Shi Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 33) pp:6097-6100
Publication Date(Web):
DOI:10.1002/anie.200902262
Co-reporter:Xiao Chen Dr.;KearyM. Engle;Dong-Hui Wang Dr.
Angewandte Chemie 2009 Volume 121( Issue 28) pp:5196-5217
Publication Date(Web):
DOI:10.1002/ange.200806273
Co-reporter:Yong-Qing Huang, Zhong-Liang Shen, Taka-aki Okamura, Yan Wang, Xiao-Feng Wang, Wei-Yin Sun, Jin-Quan Yu and Norikazu Ueyama
Dalton Transactions 2008 (Issue 2) pp:204-213
Publication Date(Web):04 Dec 2007
DOI:10.1039/B714502K
Seven new silver(I) complexes of the formula [Ag2(L)2(CF3SO3)2] (1), [Ag2(L)2(CH3SO3)2] (2) [Ag2(L)2](BF4)2 (3), [Ag3(L)2(NO3)2]NO3·5H2O (4), [Ag2(L)(NO3)2]·CH3OH (5), [Ag2(L)2](ClO4)2 (6) and [Ag3(L)2(CH3CN)3](ClO4)3 (7) have been synthesized by reactions of 1,3,5-tris(2-oxazolinyl)benzene (L) with varied silver(I) salts under different conditions. The influences of counter anions and reaction conditions on the structure of the complexes are discussed. Three complexes 1, 2 and 3 with two kinds of different 1D chain structures were obtained under the same synthetic conditions by using different silver(I) salts, and the ligand L was found to adopt bis-monodentate (1 and 2) and tris-monodentate (3) coordination modes respectively. On the other hand, by using the same silver(I) nitrate or silver(I) perchlorate but different reaction solvents, 4 and 5 or 6 and 7 were isolated respectively. Complexes 4 and 5 have different 1D chain structures, and 6 is isostructural with 3. However, 7 is a tri-nuclear, propeller-shaped M3L2 supramolecular capsule in which L adopts a cis,cis,cis-conformation, while the ligand L in 3–6 has cis,trans,trans-conformation. The results revealed that the nature of the counter anions, such as their size, coordination ability and coordination mode, and the reaction conditions all have great impact on the structure of the complexes. The complexes were also characterized by electrospray mass spectrometry. Furthermore, complex 7 exhibited modest second-harmonic-generation (SHG) efficiency.
Co-reporter:Bing-Feng Shi Dr.;Nathan Maugel;Yang-Hui Zhang Dr. Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 26) pp:4882-4886
Publication Date(Web):
DOI:10.1002/anie.200801030
Co-reporter:Bing-Feng Shi Dr.;Nathan Maugel;Yang-Hui Zhang Dr. Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 26) pp:
Publication Date(Web):
DOI:10.1002/anie.200890122
Co-reporter:Tian-Sheng Mei;Ramesh Giri;Nathan Maugel Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 28) pp:5215-5219
Publication Date(Web):
DOI:10.1002/anie.200705613
Co-reporter:Jiao-Jie Li;Tian-Sheng Mei
Angewandte Chemie International Edition 2008 Volume 47( Issue 34) pp:6452-6455
Publication Date(Web):
DOI:10.1002/anie.200802187
Co-reporter:Ramesh Giri, Nathan Maugel, Bruce M. Foxman and Jin-Quan Yu
Organometallics 2008 Volume 27(Issue 8) pp:1667-1670
Publication Date(Web):March 26, 2008
DOI:10.1021/om8000444
Pd(OAc)2-mediated dehydrogenation of an alkyl group to a double bond or a η3-allylic complex via sp3 C−H bond activation and allylic oxidation is reported. A novel redox is proposed for this double oxidation under oxidant-free conditions. A catalytic protocol using benzoquinone as the stoichiometric oxidant has also been developed for the dehydrogenation of cyclopentylcarboxamides.
Co-reporter:Bing-Feng Shi Dr.;Nathan Maugel;Yang-Hui Zhang Dr. Dr.
Angewandte Chemie 2008 Volume 120( Issue 26) pp:4960-4964
Publication Date(Web):
DOI:10.1002/ange.200801030
Co-reporter:Bing-Feng Shi Dr.;Nathan Maugel;Yang-Hui Zhang Dr. Dr.
Angewandte Chemie 2008 Volume 120( Issue 26) pp:
Publication Date(Web):
DOI:10.1002/ange.200890176
Co-reporter:Tian-Sheng Mei;Ramesh Giri;Nathan Maugel Dr.
Angewandte Chemie 2008 Volume 120( Issue 28) pp:5293-5297
Publication Date(Web):
DOI:10.1002/ange.200705613
Co-reporter:Jiao-Jie Li;Tian-Sheng Mei
Angewandte Chemie 2008 Volume 120( Issue 34) pp:6552-6555
Publication Date(Web):
DOI:10.1002/ange.200802187
Co-reporter:Yi Lu ; Dong-Hui Wang ; Keary M. Engle
Journal of the American Chemical Society () pp:
Publication Date(Web):April 1, 2010
DOI:10.1021/ja101909t
A novel Pd(II)-catalyzed ortho-C−H olefination protocol has been developed using spatially remote, unprotected tertiary, secondary, and primary alcohols as the directing groups. Mono-N-protected amino acid ligands were found to promote the reaction, and an array of olefin coupling partners could be used. When electron-deficient alkenes were used, the resulting olefinated intermediates underwent subsequent Pd(II)-catalyzed oxidative intramolecular cyclization to give the corresponding pyran products, which could be converted into ortho-alkylated alcohols under hydrogenolysis conditions. The mechanistic details of the oxidative cyclization step are discussed and situated in the context of the overall catalytic cycle.
Co-reporter:Xing-Guo Zhang ; Hui-Xiong Dai ; Masayuki Wasa
Journal of the American Chemical Society () pp:
Publication Date(Web):July 11, 2012
DOI:10.1021/ja305259n
A Pd(II)-catalyzed trifluoromethylation of ortho C–H bonds with an array of N-arylbenzamides derived from benzoic acids is reported. N-Methylformamide has been identified as a crucial promoter of C–CF3 bond formation from the Pd center. X-ray characterization of the C–H insertion intermediate has revealed a rare coordination mode of acidic amides as directing groups and the origin of their capacity in directing C–H activation.
Co-reporter:Masayuki Wasa ; Keary M. Engle ; David W. Lin ; Eun Jeong Yoo
Journal of the American Chemical Society () pp:
Publication Date(Web):November 7, 2011
DOI:10.1021/ja207607s
Systematic ligand development has led to the identification of novel mono-N-protected amino acid ligands for Pd(II)-catalyzed enantioselective C–H activation of cyclopropanes. A diverse range of organoboron reagents can be used as coupling partners, and the reaction proceeds under mild conditions. These results provide a new retrosynthetic disconnection for the construction of enantioenriched cis-substituted cyclopropanecarboxylic acids.
Co-reporter:Eun Jeong Yoo ; Masayuki Wasa
Journal of the American Chemical Society () pp:
Publication Date(Web):November 17, 2010
DOI:10.1021/ja108754f
Pd(II)-catalyzed β-C(sp3)−H carbonylation of N-arylamides under CO (1 atm) has been achieved. Following amide-directed C(sp3)−H cleavage and insertion of CO into the resulting [Pd(II)−C(sp3)] bond, intramolecular C−N reductive elimination gave the corresponding succinimides, which could be readily converted to 1,4-dicarbonyl compounds. This method was found to be effective with substrates containing α-hydrogen atoms and could be applied to effect methylene C(sp3)−H carbonylation of cyclopropanes.
Co-reporter:Ming Shang, Qian Shao, Shang-Zheng Sun, Yan-Qiao Chen, Hui Xu, Hui-Xiong Dai and Jin-Quan Yu
Chemical Science (2010-Present) 2017 - vol. 8(Issue 2) pp:NaN1473-1473
Publication Date(Web):2016/09/26
DOI:10.1039/C6SC03383K
The use of a weakly coordinating monodentate directing group for copper mediated ortho-hydroxylation and amination reactions allows for the identification of an external oxazoline ligand as a promoter.
Co-reporter:Yi Lu, Huai-Wei Wang, Jillian E. Spangler, Kai Chen, Pei-Pei Cui, Yue Zhao, Wei-Yin Sun and Jin-Quan Yu
Chemical Science (2010-Present) 2015 - vol. 6(Issue 3) pp:NaN1927-1927
Publication Date(Web):2015/01/08
DOI:10.1039/C4SC03350G
The oxidative olefination of a broad array of arenes and heteroarenes with a variety of activated and unactivated olefins has be achieved via a rhodium(III)-catalyzed C–H activation reaction. The use of an N-pentafluorophenyl benzamide directing group is crucial for achieving catalytic turnovers in the presence of air as the sole oxidant without using a co-oxidant.
Co-reporter:Yong-Qing Huang, Zhong-Liang Shen, Taka-aki Okamura, Yan Wang, Xiao-Feng Wang, Wei-Yin Sun, Jin-Quan Yu and Norikazu Ueyama
Dalton Transactions 2008(Issue 2) pp:NaN213-213
Publication Date(Web):2007/12/04
DOI:10.1039/B714502K
Seven new silver(I) complexes of the formula [Ag2(L)2(CF3SO3)2] (1), [Ag2(L)2(CH3SO3)2] (2) [Ag2(L)2](BF4)2 (3), [Ag3(L)2(NO3)2]NO3·5H2O (4), [Ag2(L)(NO3)2]·CH3OH (5), [Ag2(L)2](ClO4)2 (6) and [Ag3(L)2(CH3CN)3](ClO4)3 (7) have been synthesized by reactions of 1,3,5-tris(2-oxazolinyl)benzene (L) with varied silver(I) salts under different conditions. The influences of counter anions and reaction conditions on the structure of the complexes are discussed. Three complexes 1, 2 and 3 with two kinds of different 1D chain structures were obtained under the same synthetic conditions by using different silver(I) salts, and the ligand L was found to adopt bis-monodentate (1 and 2) and tris-monodentate (3) coordination modes respectively. On the other hand, by using the same silver(I) nitrate or silver(I) perchlorate but different reaction solvents, 4 and 5 or 6 and 7 were isolated respectively. Complexes 4 and 5 have different 1D chain structures, and 6 is isostructural with 3. However, 7 is a tri-nuclear, propeller-shaped M3L2 supramolecular capsule in which L adopts a cis,cis,cis-conformation, while the ligand L in 3–6 has cis,trans,trans-conformation. The results revealed that the nature of the counter anions, such as their size, coordination ability and coordination mode, and the reaction conditions all have great impact on the structure of the complexes. The complexes were also characterized by electrospray mass spectrometry. Furthermore, complex 7 exhibited modest second-harmonic-generation (SHG) efficiency.
Co-reporter:Ramesh Giri, Bing-Feng Shi, Keary M. Engle, Nathan Maugel and Jin-Quan Yu
Chemical Society Reviews 2009 - vol. 38(Issue 11) pp:NaN3272-3272
Publication Date(Web):2009/10/01
DOI:10.1039/B816707A
This critical review discusses historical and contemporary research in the field of transition metal-catalyzed carbon–hydrogen (C–H) bond activation through the lens of stereoselectivity. Research concerning both diastereoselectivity and enantioselectivity in C–H activation processes is examined, and the application of concepts in this area for the development of novel carbon–carbon and carbon–heteroatom bond-forming reactions is described. Throughout this review, an emphasis is placed on reactions that are (or may soon become) relevant in the realm of organic synthesis (221 references).
Co-reporter:Yi Lu, Dasheng Leow, Xisheng Wang, Keary M. Engle and Jin-Quan Yu
Chemical Science (2010-Present) 2011 - vol. 2(Issue 5) pp:NaN971-971
Publication Date(Web):2011/03/18
DOI:10.1039/C0SC00633E
A Pd(II)-catalyzed C–H carbonylation protocol of phenethyl alcohols has been developed using amino acid ligands to promote the reaction. This transformation provides an expedient route to 1-isochromanone motifs, which are common structural elements in natural products and other biologically active compounds. A concise synthesis of a histamine release inhibitor showcases the utility of this transformation.
Co-reporter:Mengchun Ye, Andrew J. F. Edmunds, James A. Morris, David Sale, Yejia Zhang and Jin-Quan Yu
Chemical Science (2010-Present) 2013 - vol. 4(Issue 6) pp:NaN2379-2379
Publication Date(Web):2013/03/18
DOI:10.1039/C3SC50184A
C3-arylated indazole and pyrazoles are privileged structural motifs in agrochemicals and pharmaceuticals. C-3 C–H arylation of (1H) indazole and pyrazole has been a significant challenge due to the poor reactivity of the C-3 position. Herein, we report a practical Pd(II)/Phen catalyst and conditions for the direct C-3 arylation of indazole and pyrazole with ArI or ArBr without using Ag additives as halide scavengers. The use of toluene, chlorobenzene, trifluoromethylbenzene and mesitylene as the solvent was found to be crucial for the selectivity and reactivity. We further demonstrate the robustness of this protocol through the first total synthesis of nigellidine hydrobromide as well as the expedient preparation of heterocycles structurally related to pesticides and drug molecules.
.jpg)
![[1,1-Biphenyl]-2-carbothioic acid, S-ethyl ester](/data/chemimg/3739400/1632163-04-2.png)
![[1,1-Biphenyl]-2-carbothioic acid, S-ethyl ester](/data/chemimg/3739400/1632163-04-2_b.png)


![Benzamide, N-[2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]phenyl]-](/data/chemimg/3721600/1340476-99-4.png)
![Benzamide, N-[2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]phenyl]-](/data/chemimg/3721600/1340476-99-4_b.png)




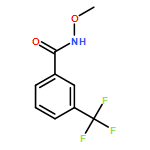



















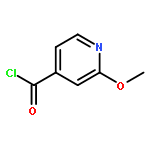

![[1,1'-Biphenyl]-4-carboxamide, N-methoxy-](/data/chemimg/105400/173171-19-2.png)
![[1,1'-Biphenyl]-4-carboxamide, N-methoxy-](/data/chemimg/105400/173171-19-2_b.png)


![Benzoic acid, 4-[(methoxyamino)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/137100/137042-75-2.png)
![Benzoic acid, 4-[(methoxyamino)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/137100/137042-75-2_b.png)










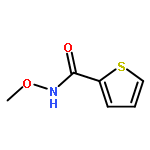

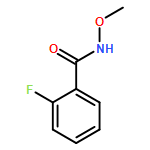
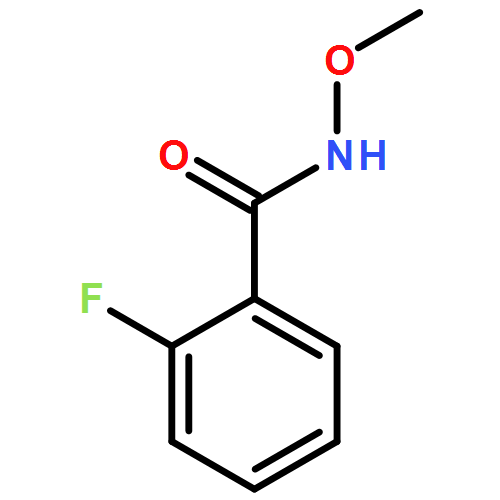














![Acetamide, N-(4'-methyl[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/76500/76472-82-7.png)
![Acetamide, N-(4'-methyl[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/76500/76472-82-7_b.png)



















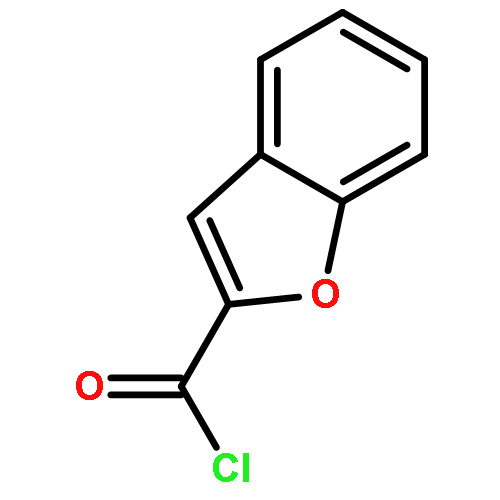


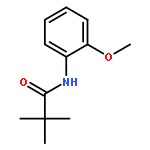
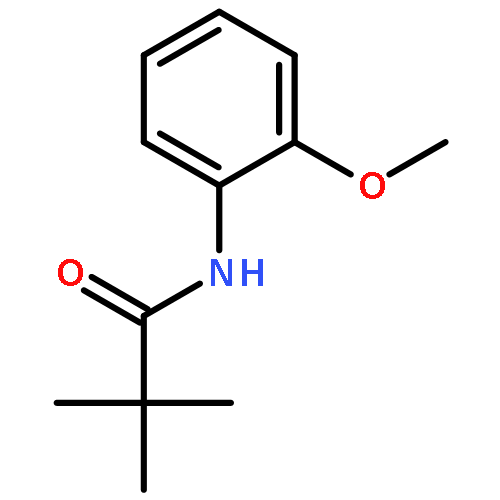
















![2-{[(4-methylphenyl)sulfonyl]amino}benzoic acid](http://img.cochemist.com/ccimg/6400/6311-23-5.png)
![2-{[(4-methylphenyl)sulfonyl]amino}benzoic acid](http://img.cochemist.com/ccimg/6400/6311-23-5_b.png)










![Propanamide,2,2-dimethyl-N-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/2000/1939-19-1.png)
![Propanamide,2,2-dimethyl-N-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/2000/1939-19-1_b.png)