Co-reporter:Quanli Gu;Yong Xia;Zhijun Yang;Carl O. Trindle;Joseph L. Knee
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 36) pp:24399-24411
Publication Date(Web):2017/09/20
DOI:10.1039/C7CP03917D
Hydrogen (H) bonds are of fundamental importance in a wide range of molecular sciences. While the study of two-center H-bonding A⋯H is well advanced, much remains to be learned in a quantitative and definitive manner for complexes with multiple H-bonds. Exemplary cases are in the category of alpha hydroxy carboxylic acids, i.e., the complexes of glycolic acid with water and formic acid. In glycolic acid, an intramolecular H-bond forms between the carboxyl group and the alpha OH group. The alpha OH stretching frequency may be affected upon complexing with water or formic acid. Direct study of glycolic acid complexes is unfortunately very difficult. However, an aromatic analogue, 9-hydroxy-9-fluorene carboxylic acid (9HFCA), permits detailed and accurate gas phase spectroscopic studies. Since computational analysis establishes that H-bonding is very similar from glycolic acid complexes to 9HFCA complexes, direct studies on 9HFCA complexes by laser spectroscopy also deduce new and subtle information for glycolic acid complexes in terms of molecular structures, binding strength, and collective effects of multiple H-bonds associated with anti-cooperativity and cooperativity.
Co-reporter:Quanli Gu;Dan Shen;Zhen Tang;Wei Wu;Yong Xia;Zhijun Yang;Carl O. Trindle
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 22) pp:14238-14247
Publication Date(Web):2017/06/07
DOI:10.1039/C7CP02234D
The binding strength and collective effects of multiple H-bonds in the glycolic acid–water dimer were studied in comparison to the aromatic analog, 9-hydroxy-9-fluorene carboxylic acid (9HFCA). Quantitative analysis by the generalized Kohn–Sham energy decomposition analysis shows that the energy difference in each specific physical interaction, from a glycolic acid–water dimer to a 9HFCA–water dimer, is small and amounts to less than 5% of the binding energy of the 9HFCA–water dimer. Extensive comparison of further, similar H-bonded complexes with widely varying binding strengths reinforces their excellent analogy in that the fluorene group acts as a non-interfering spectator for intermolecular H-bonding interactions. With reference to the spectroscopic measurement on the 9HFCA–water dimer (8.51 ± 0.09 kcal mol−1), the binding energy of the glycolic acid–water dimer is estimated to be 8.51 ± 0.31 kcal mol−1, a much better accuracy than previous reports. Furthermore, correlating the infrared spectra of 9HFCA H-bonded complexes provides a circumstantial probing of the existence and consequences of cooperative and anti-cooperative behaviors in the glycolic acid–water dimer. Our studies point to the interesting H-bonding phenomena in the glycolic acid–water dimer, which may inspire challenging experiments in future.
Co-reporter:Chen Zhou, Yang Zhang, Xiping Gong, Fuming Ying, Peifeng SuWei Wu
Journal of Chemical Theory and Computation 2017 Volume 13(Issue 2) pp:
Publication Date(Web):December 19, 2016
DOI:10.1021/acs.jctc.6b01144
In this work, a valence bond type multireference density functional theory (MRDFT) method, called the Hamiltonian matrix correction based density functional valence bond method (hc-DFVB), is presented. In hc-DFVB, the static electronic correlation is considered by the valence bond self-consistent field (VBSCF) strategy, while the dynamic correlation energy is taken into account by Kohn–Sham density functional theory (KS-DFT). Different from our previous version of DFVB (J. Chem. Theory Comput. 2012, 8, 1608), hc-DFVB corrects the dynamic correlation energy with a Hamiltonian correction matrix, improving the functional adaptability and computational accuracy. The method was tested for various physical and chemical properties, including spectroscopic constants, bond dissociation energies, reaction barriers, and singlet–triplet gaps. The accuracy of hc-DFVB matches that of KS-DFT and high level molecular orbital (MO) methods quite well. Furthermore, hc-DFVB keeps the advantages of VB methods, which are able to provide clear interpretations and chemical insights with compact wave functions.
Co-reporter:Zongyuan Liu;Carl O. Trindle;Quanli Gu;Wei Wu
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 37) pp:25260-25269
Publication Date(Web):2017/09/27
DOI:10.1039/C7CP03491A
The neutral and cationic forms of tryptamine–water dimer present a variety of noncovalent interactions. To characterize these interactions, a series of complementary methods, including the quantum theory of atoms in molecules, noncovalent interaction plots, natural bond orbital analysis, and energy decomposition analysis, were used. For the first time, the existence of the three intermolecular H-bonds in the conformer-locked tryptamine–water dimer A–H2O are identified, highlighting a single water's role as one proton donor and two proton acceptors as it binds to tryptamine. Furthermore, upon threshold ionization of the A–H2O dimer, the network of the three intermolecular H-bonds is indeed preserved while the individual H-bonds’ binding strengths are subject to change; this is attributed to the existence of the optically accessible minimum energy isomer A+–H2O in the cation. In addition, it is found that the global minimum energy isomer H+–H2O contains a single intermolecular H-bond, but is more stable, by ca. 3 kcal mol−1, than the local minimum energy isomer A+–H2O; this is due to the stronger intramolecular interaction of H+–H2O as opposed to A+–H2O.
Co-reporter:Peifeng Su;Hongjiang Chen;Wei Wu
Science China Chemistry 2016 Volume 59( Issue 8) pp:1025-1032
Publication Date(Web):2016 August
DOI:10.1007/s11426-016-0007-2
In this work, the intra-EDA method, which is a recently developed energy decomposition analysis scheme for intramolecular non-covalent interaction is extended from gas phase to solvated environment. It is the first analysis scheme that performs analysis for intramolecular interaction in solution. By fragmentation scheme, a molecule is divided into intramolecular interacting fragments and environmental fragments via single bond homolysis breaking. The solvent effect is taken into account by implicit solvation model. Intramolecular interaction free energy is estimated as the separated treatment of inter-fragment interactions in dielectric environment. The analysis results highlight the importance of solvent effects to intramolecular non-covalent interaction.
Co-reporter:Xin Chang, Yang Zhang, Xinzhen Weng, Peifeng Su, Wei Wu, and Yirong Mo
The Journal of Physical Chemistry A 2016 Volume 120(Issue 17) pp:2749-2756
Publication Date(Web):April 13, 2016
DOI:10.1021/acs.jpca.6b02245
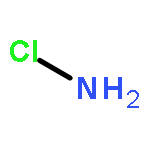
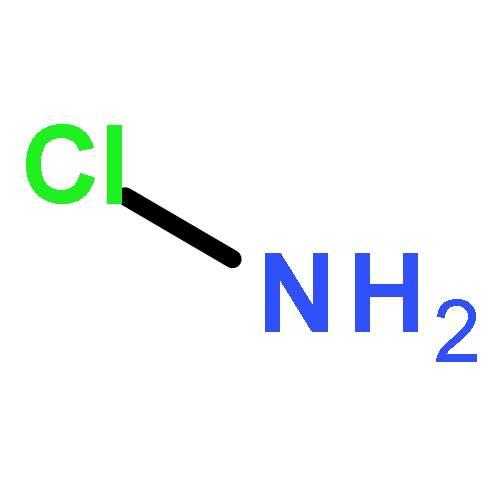
Both proper, red-shifting and improper, blue-shifting hydrogen bonds have been well-recognized with enormous experimental and computational studies. The current consensus is that there is no difference in nature between these two kinds of hydrogen bonds, where the electrostatic interaction dominates. Since most if not all the computational studies are based on molecular orbital theory, it would be interesting to gain insight into the hydrogen bonds with modern valence bond (VB) theory. In this work, we performed ab initio VBSCF computations on a series of hydrogen-bonding systems, where the sole hydrogen bond donor CF3H interacts with ten hydrogen bond acceptors Y (═NH2CH3, NH3, NH2Cl, OH–, H2O, CH3OH, (CH3)2O, F–, HF, or CH3F). This series includes four red-shifting and six blue-shifting hydrogen bonds. Consistent with existing findings in literature, VB-based energy decomposition analyses show that electrostatic interaction plays the dominating role and polarization plays the secondary role in all these hydrogen-bonding systems, and the charge transfer interaction, which denotes the hyperconjugation effect, contributes only slightly to the total interaction energy. As VB theory describes any real chemical bond in terms of pure covalent and ionic structures, our fragment interaction analysis reveals that with the approaching of a hydrogen bond acceptor Y, the covalent state of the F3C–H bond tends to blue-shift, due to the strong repulsion between the hydrogen atom and Y. In contrast, the ionic state F3C– H+ leads to the red-shifting of the C–H vibrational frequency, owing to the attraction between the proton and Y. Thus, the relative weights of the covalent and ionic structures essentially determine the direction of frequency change. Indeed, we find the correlation between the structural weights and vibrational frequency changes.
Co-reporter:Peifeng Su, Zuochang Chen, Wei Wu
Chemical Physics Letters 2015 Volume 635() pp:250-256
Publication Date(Web):16 August 2015
DOI:10.1016/j.cplett.2015.06.078
Highlights
- •
Our scheme is a combination of fragmentation and energy decomposition analysis.
- •
Intramolecular interaction can be modeled as intermolecular interaction.
- •
Total interaction energy is analyzed by energy decomposition analysis.
Co-reporter:Jing Huang;FuMing Ying;Wei Wu
Science China Chemistry 2014 Volume 57( Issue 10) pp:1409-1417
Publication Date(Web):2014 October
DOI:10.1007/s11426-014-5192-x
In this paper, a combined QM/MM/PCM approach, named VBEFP/PCM, is presented for ab initio VB study with a solvent effect incorporated. In VBEFP/PCM, both short-range and long-range solvent effects are taken into account by effective fragment potential (EFP) and polarizable continuum model (PCM), respectively, while the solute molecules are described by valence bond (VB) wave function. Furthermore, VBEFP/PCM, along with VBPCM and VBEFP, is employed for the n→π* vertical excitation of formaldehyde and acetone molecules in aqueous solution. The computational results show that VBEFP/PCM can provide the appropriate solvent shifts, whereas VBPCM underestimates the solvent shifts due to its lack of short-range solvent effect. The VBEFP results strongly rely upon the description of the short-range solvent effect. To explore the role of the solute’s electronic structure in the solvent shift, resonance energy analysis during the excitation is performed. It was found that the solute’s electronic polarization plays the most important role in the solvent shift. The resonance controls the variation of the solute’s wave function during the n→* vertical excitation, which leads to the blue solvent shifts.
Co-reporter:Peifeng Su, Zhen Jiang, Zuochang Chen, and Wei Wu
The Journal of Physical Chemistry A 2014 Volume 118(Issue 13) pp:2531-2542
Publication Date(Web):March 10, 2014
DOI:10.1021/jp500405s
In this paper, a new energy decomposition analysis scheme based on the generalized Kohn–Sham (GKS) and the localized molecular orbital energy decomposition analysis (LMO-EDA) scheme, named GKS-EDA, is proposed. The GKS-EDA scheme has a wide range of DFT functional adaptability compared to LMO-EDA. In the GKS-EDA scheme, the exchange, repulsion, and polarization terms are determined by DFT orbitals; the correlation term is defined as the difference of the GKS correlation energy from monomers to supermolecule. Using the new definition, the GKS-EDA scheme avoids the error of LMO-EDA which comes from the separated treatment of EX and EC functionals. The scheme can perform analysis both in the gas and in the condensed phases with most of the popular DFT functionals, including LDA, GGA, meta-GGA, hybrid GGA/meta-GGA, double hybrid, range-separated (long-range correction), and dispersion correction. By the GKS-EDA scheme, the DFT functionals assessment for hydrogen bonding, vdW interaction, symmetric radical cation, charge-transfer, and metal–ligand interaction is performed.
Co-reporter:Xin Chang, Peifeng Su, Wei Wu
Chemical Physics Letters 2014 s 610–611() pp: 246-250
Publication Date(Web):
DOI:10.1016/j.cplett.2014.07.036
Co-reporter:Fuming Ying, Xin Chang, Peifeng Su, and Wei Wu
The Journal of Physical Chemistry A 2012 Volume 116(Issue 7) pp:1846-1853
Publication Date(Web):January 25, 2012
DOI:10.1021/jp211314j
An ab initio explicit solvation valence bond (VB) method, called VBEFP, is presented. The VBEFP method is one type of QM/MM approach in which the QM part of system is treated within the ab initio valence bond scheme and the solvent water molecules are accounted by the effective fragment potential (EFP) method, which is a polarized force field approach developed by Gordon et al. ( J. Chem. Phys. 1996, 105, 1968). This hybrid method enables one to take the first-solvation shell and heterogeneous solvation effects into account explicitly with VB wave function. Therefore, the nature of chemical bonding and the mechanism of chemical reactions with explicit solvent environments can be explored at the ab inito VB level. In this paper, the hydrated metal–ligand complexes [M2+L](H2O)n (M2+: Mg2+, Zn2+; L: NH3, CH2O) are studied by the VBEFP method. Resonance energy and bond order are computed, and the influence of the solvent coordination and hydrogen bonding to the metal–ligand bonding are explored in the paper.
Co-reporter:Quanli Gu, Dan Shen, Zhen Tang, Wei Wu, Peifeng Su, Yong Xia, Zhijun Yang and Carl O. Trindle
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 22) pp:NaN14247-14247
Publication Date(Web):2017/05/03
DOI:10.1039/C7CP02234D
The binding strength and collective effects of multiple H-bonds in the glycolic acid–water dimer were studied in comparison to the aromatic analog, 9-hydroxy-9-fluorene carboxylic acid (9HFCA). Quantitative analysis by the generalized Kohn–Sham energy decomposition analysis shows that the energy difference in each specific physical interaction, from a glycolic acid–water dimer to a 9HFCA–water dimer, is small and amounts to less than 5% of the binding energy of the 9HFCA–water dimer. Extensive comparison of further, similar H-bonded complexes with widely varying binding strengths reinforces their excellent analogy in that the fluorene group acts as a non-interfering spectator for intermolecular H-bonding interactions. With reference to the spectroscopic measurement on the 9HFCA–water dimer (8.51 ± 0.09 kcal mol−1), the binding energy of the glycolic acid–water dimer is estimated to be 8.51 ± 0.31 kcal mol−1, a much better accuracy than previous reports. Furthermore, correlating the infrared spectra of 9HFCA H-bonded complexes provides a circumstantial probing of the existence and consequences of cooperative and anti-cooperative behaviors in the glycolic acid–water dimer. Our studies point to the interesting H-bonding phenomena in the glycolic acid–water dimer, which may inspire challenging experiments in future.