Co-reporter:Meiheng Lu;Yunfan Yang;Tianshu Chu
Theoretical Chemistry Accounts 2017 Volume 136( Issue 5) pp:
Publication Date(Web):2017 May
DOI:10.1007/s00214-017-2088-9
The excited-state double-proton transfer mechanism of 2,5-bis(benzoxazol-2-yl)thiophene-3,4-diol has been theoretically investigated based on the methods of density functional theory and time-dependent density functional theory. Geometric structure comparison and infrared vibrational spectra analysis confirm that the strengthening of the intramolecular hydrogen bond in the first excited state (S1) has facilitated the proton transfer process. The frontier molecular orbitals analysis illustrates that the nature of the hydrogen bond enhancement lies in the charge redistribution upon photo-excitation, which is further confirmed by NBO charge analysis quantitatively. The reduced dimensionality (two-dimensional) potential energy surfaces have been scanned for both the ground state (S0) and the first electronic excited state to reveal the mechanism and pathway of the two protons being transferred. Compared with the concerted path of proton transfer, the stepwise path with one proton being transferred first and then a second one followed exhibits a relatively mild energy barrier and seems to be more rational and favorable.
Co-reporter:Meiheng Lu;Yunfan Yang;Tianshu Chu
Theoretical Chemistry Accounts 2017 Volume 136( Issue 5) pp:
Publication Date(Web):2017 May
DOI:10.1007/s00214-017-2088-9
The excited-state double-proton transfer mechanism of 2,5-bis(benzoxazol-2-yl)thiophene-3,4-diol has been theoretically investigated based on the methods of density functional theory and time-dependent density functional theory. Geometric structure comparison and infrared vibrational spectra analysis confirm that the strengthening of the intramolecular hydrogen bond in the first excited state (S1) has facilitated the proton transfer process. The frontier molecular orbitals analysis illustrates that the nature of the hydrogen bond enhancement lies in the charge redistribution upon photo-excitation, which is further confirmed by NBO charge analysis quantitatively. The reduced dimensionality (two-dimensional) potential energy surfaces have been scanned for both the ground state (S0) and the first electronic excited state to reveal the mechanism and pathway of the two protons being transferred. Compared with the concerted path of proton transfer, the stepwise path with one proton being transferred first and then a second one followed exhibits a relatively mild energy barrier and seems to be more rational and favorable.
Co-reporter:Tian-shu Chu, Rui Lü, Bai-tong Liu
Biosensors and Bioelectronics 2016 Volume 86() pp:643-655
Publication Date(Web):15 December 2016
DOI:10.1016/j.bios.2016.07.039
•Recent advances in developing redox-responsive reversible NIR biosensors.•Spectroscopic property and fluorescence sensing mechanism.•Design strategy for redox-responsive reversible NIR biosensors.•Applications of NIR biosensors in in vitro and in vivo fluorescence imaging.•Successful monitoring of multiple redox cycles in living biological systems.Reactive oxygen species (ROS) and changes in their redox cycles have great therapeutic potential for treating serious redox-related human diseases such as acute and chronic inflammation, diabetes, cancer and neurodegenerative disorders. This article presents a survey of the recently (2011–2016) developed NIR small-molecule biosensors for reversibly monitoring oxidation and reduction events in living cells and small animals through in vitro/in vivo fluorescence imaging. Emission and absorption profile, design strategy and fluorescence sensing mechanism, ROS selectivity and sensitivity, reversibility, ability of subcellular location and cytotoxicity are discussed for the NIR small-molecule biosensors capable of quantitatively, continuously and reversibly detecting transient ROS burst and redox changes at cellular level.
Co-reporter:Runze Liu, Yinghe Zhao and Tianshu Chu
Chemical Communications 2015 vol. 51(Issue 12) pp:2429-2432
Publication Date(Web):01 Jan 2015
DOI:10.1039/C4CC09424G
We studied the reaction mechanism of di-n-butylmagnesium decomposing into MgH2 in cyclohexane, and found a new route easier than famous β-hydride elimination. Further, we explored the dynamic behavior of graphene nano-flakes and MgH2 in cyclohexane, and gained new insights for efficient hydrogen storage material preparation.
Co-reporter:Jun-Sheng Chen, Feng-Jiao Zhao, Yang Yang and Tian-Shu Chu
RSC Advances 2015 vol. 5(Issue 46) pp:36279-36287
Publication Date(Web):14 Apr 2015
DOI:10.1039/C5RA04005A
In this work, spectroscopic techniques and quantum chemistry calculations were used to investigate the photophysical properties of 2-ureido-4[1H]-pyrimidinone (UPy) systems in two different solvents of DMSO and DCM. The investigations were carried out on the three UPy systems (AnUP, NaUP and UPNa) with two different choromophores (i.e., anthracene and naphthalene) located at the head and the tail part of the UPy module. In DCM the UPy systems exist as the Keto-2 form that self assembles into a dimer through quadruple H-B arrays AADD–DDAA (solute–solute hydrogen bonds). In DMSO the UPy systems exist in the Keto-1 form which then forms a hydrogen bond complex with the solvent through solute–solvent interaction. The differences in excited state hydrogen bond dynamics and in configuration dynamical processes, account for the measured different fluorescence lifetimes and fluorescence quantum yields (FQY, ΦF) of the UPy systems studied in this work.
Co-reporter:Jun-Sheng Chen, Pan-Wang Zhou, Li Zhao and Tian-Shu Chu
RSC Advances 2014 vol. 4(Issue 1) pp:254-259
Publication Date(Web):07 Nov 2013
DOI:10.1039/C3RA44900A
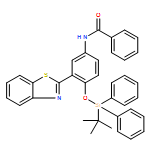
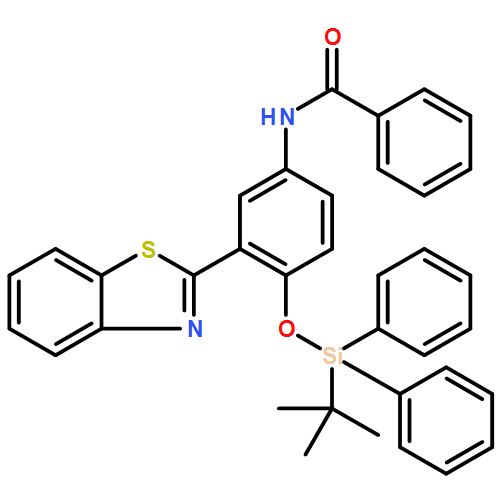
The sensing mechanism of the aqueous fluoride chemosensor N-(3-(benzo[d]thiazol-2-yl)-4-(tert-butyldiphenyl silyloxy)phenyl)-benzamide (BTTPB) has been studied in detail by DFT/TDDFT methods. The desilylation reaction which has a moderate transition barrier of 17.6 kcal mol−1 and the excited state intramolecular proton transfer (ESIPT) of the desilylation reaction product (3-BTHPB) work together for the fluorescent sensing mechanism. The constructed potential energy curves among the optimized 3-BTHPB (enol form) and 3-BTHPB-e (keto form) geometries on the S0 and S1 states, indicated that the ESIPT is a low barrier process (0.1 kcal mol−1), and the energies of the optimized geometries showed that the ESIPT process is exothermic. The calculated vertical excitation energies in the ground state and the first singlet excited state reproduced the experimental UV-Vis absorbance and fluorescence emission spectra well.
Co-reporter:Minghu Yuan, Tianshu Chu
Chemical Physics 2014 Volume 435() pp:9-13
Publication Date(Web):19 May 2014
DOI:10.1016/j.chemphys.2014.02.020
Highlights
- •
We present a 3D quantum approach for studying the atom–laser interaction.
- •
Sine-DVR and split-operator propagator are used for economizing computational time.
- •
Our calculated electron spectrum has good agreement with experimental result.
- •
The ATI spectra have studied in linearly and circularly polarized lights.
Co-reporter:Jun-Sheng Chen, Ming-Hu Yuan, Jia-Pei Wang, Yang Yang, and Tian-Shu Chu
The Journal of Physical Chemistry A 2014 Volume 118(Issue 39) pp:8986-8995
Publication Date(Web):June 4, 2014
DOI:10.1021/jp501946n
The biothiols sensing mechanism of (E)-7-(diethylamino)-3-(2-nitrovinyl)-2H-chromen-2-one (DCO) has been investigated using the density functional theory (DFT) and time-dependent DFT methods. The theoretical results indicate that the excited-state intermolecular hydrogen bonding (H–B) plays an important role for the biothiols sensing mechanism of the fluorescence sensor DCO. Multiple H–B interaction sites exist in DCO and in its Michael addition product DCOT, which then induce the formation of the H–B complexes with water molecules, DCOH2 and DCOTH4. In the first excited state, the intermolecular H–Bs between water molecule and DCO in DCOH2 are cooperatively and generally strengthened and thus induced the weak fluorescence emission of DCO, while the cooperative H–Bs between water molecule and DCOT in DCOTH4 are overall weakened and thus responsible for the enhanced fluorescence emission of DCOT. Moreover, the theoretical results suggest that the blue shift of the UV–Vis absorption spectrum of DCOT can be attributed to the relatively weak excited-state intramolecular charge transfer in DCOT compared to DCO.
Co-reporter:Dandan Wang, Rui Lü, Minghu Yuan, Aiping Fu, Tianshu Chu
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2014 Volume 125() pp:131-137
Publication Date(Web):5 May 2014
DOI:10.1016/j.saa.2014.01.094
•A DFT/TD-DFT investigation on cooperation effect of hydrogen bonding dynamics.•The thiazolidinedione derivative A and its hydrogen-bonded complexes in DMF.•The strengthening trend of the hydrogen bonding OH⋯OC.•The weakening trend of the hydrogen bonding NH⋯OC.•The cooperation effect caused a blue shift of 6 nm in the electronic spectrum.The time-dependent density functional theory (TDDFT) method has been applied to investigate the thiazolidinedione (TZD) derivative A and its hydrogen-bonded complexes with dimethylformamide (DMF) (A-DMF and A-2DMF). The calculation results showed that the excited-state hydrogen bondings of OH⋯OC and NH⋯OC are strengthened and weakened in the hydrogen-bonded trimer A-2DMF, and their cooperation effect caused a blue shift in the electronic spectrum of A-2DMF. This modulation mechanism of the hydrogen-bond strengthening and weakening and its role in influencing the spectroscopy property of the TZD derivative A in DMF have been analyzed and showed in details.Graphical abstract
Co-reporter:Dandan Wang, Rui Lü, Minghu Yuan, Junsheng Chen, Liqiang Feng, Aiping Fu, Fenghui Tian, António J.C. Varandas, Tianshu Chu
Chemical Physics Letters 2014 s 610–611() pp: 179-185
Publication Date(Web):
DOI:10.1016/j.cplett.2014.07.012
Co-reporter:Jun-Sheng Chen, Pan-Wang Zhou, Song-Qiu Yang, Ai-Ping Fu and Tian-Shu Chu
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 38) pp:16183-16189
Publication Date(Web):08 Aug 2013
DOI:10.1039/C3CP51482J
Our density functional theory (DFT)/time-dependent DFT calculations for the fluoride anion sensor, 5,7-dibromo-8-tert-butyldimethylsilyloxy-2-methylquinoline (DBM), suggested a different sensing mechanism from the experimentally proposed one (Chem. Commun., 2011, 47, 7098). Instead of the formation of fluoride–hydrogen-bond complex (DBMOHF) and excited-state proton transfer mechanism, the theoretical results predicted a sensing mechanism based on desilylation reaction and intramolecular charge transfer (ICT). The fluoride anion reacted with DBM and formed an anion (DBMO), with the ICT causing a red shift in the absorbance and emission spectra of the latter. The calculated vertical excitation energies in the ground and first excited states of both DBM and DBMO, as well as the calculated 1H NMR spectra, significantly reproduced the experimental measurements, providing additional proofs for our proposed sensing mechanism for DBM.
Co-reporter:Wenliang Li, Wenwu Xu, Tianshu Chu
Computational and Theoretical Chemistry 2013 Volume 1004() pp:18-21
Publication Date(Web):15 January 2013
DOI:10.1016/j.comptc.2012.10.017
We have demonstrated the use of the multi-configuration time-dependent Hartree–Fock (MCTDHF) method to compute total ionization probabilities of a 3-dimensional (3D) model helium atom interacting with a strong laser field. The time dependence and the intensity dependence of the total ionization probabilities of helium are investigated by using the MCTDHF method. The MCTDHF results are consistent with the previous theoretical expectations by directly solving the time-dependent Schrödinger equation.Graphical abstractHighlights► We demonstrated the use of the MCTDHF method to compute total ionization of helium. ► The intensity dependence of total ionization agrees well with previous theoretical results. ► The time-dependent total ionization probabilities are presented. ► A validation calculation of the MCTDHF method.
Co-reporter:Jun-Sheng Chen, Pan-Wang Zhou, Guang-Yue Li, Tian-Shu Chu, and Guo-Zhong He
The Journal of Physical Chemistry B 2013 Volume 117(Issue 17) pp:5212-5221
Publication Date(Web):April 11, 2013
DOI:10.1021/jp4017757
The fluoride anion sensing mechanism of 6-methyl-5-(9-methylene-anthracene)-(2-butylureido-4[1H]-pyrimidinone) (AnUP) has been investigated using the DFT/TDDFT method. The theoretical results indicate that the proton of the N3–H3 group in pyrimidine moiety is captured by the added fluoride anion and then deprotonated. The calculated vertical excitation energies of AnUP-dimer and its deprotonated form agree well with the experimental results. The molecular orbital analysis demonstrates that the first excited state (S1) of AnUP-dimer is a local excited state with a π–π* transition, whereas for the deprotonated form, S1 is a completely charge-separation state and is responsible for the photoinduced electron transfer (PET) process. The PET process from anthracene to the pyrimidine moiety leads to the fluorescence quenching.
Co-reporter:Liqiang Feng, Tianshu Chu
Journal of Electron Spectroscopy and Related Phenomena 2012 Volume 185(Issue 11) pp:458-465
Publication Date(Web):November 2012
DOI:10.1016/j.elspec.2012.09.002
In this paper, we study the issue of single quantum path control and its role in attosecond pulse generation. By carrying out the time-dependent Schrödinger equation analysis for the harmonic emission from a single He atom irradiated by the two-color laser field, consisting of a short 800 fundamental chirped pulse and a subharmonic 800–2400 nm control pulse, we find that the most favorable condition for attosecond generation is at the fundamental chirp parameter β = 4.55 together with the zero-phase 2000 nm control pulse, in which the single quantum path (short quantum path) is selected to contribute to the harmonic spectrum exhibiting an ultrabroad supercontinuum of a 337 eV bandwidth. Finally, an isolated attosecond pulse as short as 39 as is thus generated directly.Highlights► HHG spectra and attosecond pulse generation from a model He atom. ► Two-color laser field of a chirped fundamental pulse and a subharmonics control pulse. ► Single quantum path selection by β = 4.55 chirp pulse and the zero-phase 2000 nm control pulse. ► Formation of 337 eV supercontinuum region and generation of 39 as pulse.
Co-reporter:Liqiang Feng, Tianshu Chu
Chemical Physics 2012 Volume 405() pp:26-31
Publication Date(Web):11 September 2012
DOI:10.1016/j.chemphys.2012.06.001
Abstract
Quantum path control on the harmonic emission from a model Ne atom has been investigated within the scheme where a terahertz (THz) controlling pulse is added to a 5 fs/1200 nm fundamental chirp pulse. It has been found that, with the optimal THz field, the short quantum path has been selected and the supercontinuum region of the harmonic spectra has been extended from 357 eV to 656 eV with less modulated structures. The proper superposition of harmonics without any phase compensation produces a single isolated 38 as attosecond pulse.
Co-reporter:Liqiang Feng, Tianshu Chu
Journal of Electron Spectroscopy and Related Phenomena 2012 Volume 185(1–2) pp:39-46
Publication Date(Web):March 2012
DOI:10.1016/j.elspec.2011.11.004
In this paper, we theoretically investigate the delay time effect on the high-order harmonic generation (HHG) when a model Ne atom is exposed to a two-color time delayed pulse, consisting of a 5fs/800 nm fundamental field and a 20fs/2000 nm controlling field. It shows that the HHG spectra are strongly sensitive to the delay time between the two laser fields, in particular, for the zero carrier-envelope phase (CEP) φ case (corresponding to the 800 nm fundamental field), the maximum cutoff energy has been achieved at zero delay time. However, with the introduction of the CEP (φ = 180°), the delay effect on HHG is changed, exhibiting a ‘U’ structure harmonic emission from −1 T to 1 T. In addition, the combinations of different controlling pulse frequencies and pulse intensities have also been considered, showing the similar results as the original controlling field case, but with some characteristics. Finally, by properly superposing the optimal harmonic spectrum, an isolated 45as pulse is generated without phase compensation.Highlights► Investigation of HHG spectra and single isolated attosecond pulse generation. ► Irradiation from a model Ne atom by two-color time delayed pulse. ► Observation of time delay effect and relative phase effect. ► Revelation of the optimal condition for generating isolated attosecond pulse. ► Generation of a single isolated attosecond pulse of 45as.
Co-reporter:Li-Qiang Feng
Journal of Molecular Modeling 2012 Volume 18( Issue 12) pp:5097-5106
Publication Date(Web):2012 December
DOI:10.1007/s00894-012-1511-3
The ionization and the dissociation of the diatomic molecular ion H2+ have been investigated within a scheme where a noise field is added to an intense infrared laser field. The results show that both the ionization and the dissociation probabilities are enhanced with the introduction of the additional noise (the Gaussian white noise or the color noise) field. Further, by tuning the noise intensity and the delay time between the laser and the noise, a stochastic resonancelike curve is observed for the ionization or the dissociation dynamics, showing the existence of an optimal noise intensity and delay time for the given laser field.
Co-reporter:Ping Song, Ai-Hua Gao, Pan-Wang Zhou, and Tian-Shu Chu
The Journal of Physical Chemistry A 2012 Volume 116(Issue 22) pp:5392-5397
Publication Date(Web):May 16, 2012
DOI:10.1021/jp302535m
DFT and TDDFT methods have been performed to investigate the photoisomerization effect for dithiazolylarylene on solution. The weak S···N interaction and CH···N hydrogen bond restrain the rotation of the side-chain thiazolyl ring in open-isomer 1a, the higher stability of which prefers to show a high quantum yield of photoisomerization. The calculated UV–Vis spectrum at around 320 nm for open-isomer 1a is bathochromically shifted to 647 nm for closed-isomer 1b, in excellent agreement with the experimental photochromic phenomenon. The electron transition in ECD (electron circular dichroism) spectra for closed-isomer 1b with two chiral carbon atoms is dominated by ICT (intramolecular charge transition) and LE (local excitation) corresponding to one positive (440 nm) and one negative Cotton effect (650 nm), respectively, where the two chiral carbon atoms play a slight role in these transitions. The PES in the S1 and S0 states, respectively, indicates that the cyclization reaction from open-isomer 1a to closed-isomer 1b is allowed in the photoexcited state with high-conversion quantum efficiency, while it is forbidden in the thermodynamic process. In addition, the second-order nonlinear optical response for closed-isomer 1b is nearly six times larger than that for open-isomer 1a. It is also confirmed that the photoirradiation evokes the photoisomerization character to show dramatic difference in the second-order NLO response, which can be applied to designing photochromic materials and reversible NLO switches.
Co-reporter:Guang-Yue Li and Tianshu Chu
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 46) pp:20766-20771
Publication Date(Web):14 Oct 2011
DOI:10.1039/C1CP21470E
The fluoride-sensing mechanism of the sensor 2-(2′-phenylurea-phenyl)benzoxazole (PUBO) has been investigated by means of the TD-DFT method. The present theoretical study indicates that there is an excited-state intramolecular proton transfer (ESIPT) process in the sensor PUBO. The added fluoride anion could capture the proton in the free N–H moiety instead of the hydrogen-bonding one. The experimental UV/Vis and fluorescence spectra (J. Org. Chem. 2007, 72, 62) are well reproduced by the calculated vertical excitation energies in the ground state and the first singlet excited state. For example, the calculated emission wavelength of PUBO at 534 nm is very close to the fluorescence band at 554 nm. Furthermore, we theoretically confirmed that the added fluoride anions could inhibit the ESIPT process in PUBO. But different from the classical ESIPT-inhibition mechanism, the ESIPT process in the sensor PUBO is inhibited by the high energy barrier of its deprotonated form rather than by the absence of the transferred proton.
Co-reporter:Kun Zhao, Tianshu Chu
Chemical Physics Letters 2011 Volume 511(1–3) pp:166-171
Publication Date(Web):26 July 2011
DOI:10.1016/j.cplett.2011.06.005
Abstract
We discuss the possibility of using the frequency-chirping technique to shorten the duration of the generated single attosecond pulse (SAP) by a two-color laser field of 800 and 1600 nm with few-cycle pulses. By adopting various combinations of the two frequency-chirped laser fields in our numerical simulation of ionizing He atom, we demonstrate that the best possible condition to obtain the shortest SAP is using the same chirping in both the fundamental and the half-harmonic laser fields without any phase effect and any delay time. There is a maximum increment of about 40 eV in the bandwidth of the XUV super-continuum in the cutoff (the second plateau) region. A single isolated attosecond pulse of 48 as can be generated that is further reduced to 9.7 as by phase compensation.
Co-reporter:Tian Shu Chu, Yun Bo Duan, Shu Ping Yuan, António J.C. Varandas
Chemical Physics Letters 2007 Volume 444(4–6) pp:351-354
Publication Date(Web):27 August 2007
DOI:10.1016/j.cplett.2007.07.057
Accurate time-dependent wave packet calculations have been performed for the isotopic N(2D) + D2 reaction on the recent reported 12A″ DMBE (double many body expansion) potential energy surface of the NH2 system. The total reaction probabilities for total angular momentum J up to 75 have been calculated to get the converged integral cross-section over a collision energy range of 0.0–1.0 eV. Then, the ground-state thermal rate constant has been derived from the integral cross-section and the intermolecular isotope effect is further discussed.The ground-state thermal rate constant and the intermolecular isotope effect for N(2D) + D2, obtained from the present time-dependent wave packet calculation on the 12A″ DMBE potential energy surface, along with the experimental and theoretical results. As can be seen, a qualitative agreement has been achieved.
Co-reporter:Jun-Sheng Chen, Pan-Wang Zhou, Song-Qiu Yang, Ai-Ping Fu and Tian-Shu Chu
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 38) pp:NaN16189-16189
Publication Date(Web):2013/08/08
DOI:10.1039/C3CP51482J
Our density functional theory (DFT)/time-dependent DFT calculations for the fluoride anion sensor, 5,7-dibromo-8-tert-butyldimethylsilyloxy-2-methylquinoline (DBM), suggested a different sensing mechanism from the experimentally proposed one (Chem. Commun., 2011, 47, 7098). Instead of the formation of fluoride–hydrogen-bond complex (DBMOHF) and excited-state proton transfer mechanism, the theoretical results predicted a sensing mechanism based on desilylation reaction and intramolecular charge transfer (ICT). The fluoride anion reacted with DBM and formed an anion (DBMO), with the ICT causing a red shift in the absorbance and emission spectra of the latter. The calculated vertical excitation energies in the ground and first excited states of both DBM and DBMO, as well as the calculated 1H NMR spectra, significantly reproduced the experimental measurements, providing additional proofs for our proposed sensing mechanism for DBM.
Co-reporter:Guang-Yue Li and Tianshu Chu
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 46) pp:NaN20771-20771
Publication Date(Web):2011/10/14
DOI:10.1039/C1CP21470E
The fluoride-sensing mechanism of the sensor 2-(2′-phenylurea-phenyl)benzoxazole (PUBO) has been investigated by means of the TD-DFT method. The present theoretical study indicates that there is an excited-state intramolecular proton transfer (ESIPT) process in the sensor PUBO. The added fluoride anion could capture the proton in the free N–H moiety instead of the hydrogen-bonding one. The experimental UV/Vis and fluorescence spectra (J. Org. Chem. 2007, 72, 62) are well reproduced by the calculated vertical excitation energies in the ground state and the first singlet excited state. For example, the calculated emission wavelength of PUBO at 534 nm is very close to the fluorescence band at 554 nm. Furthermore, we theoretically confirmed that the added fluoride anions could inhibit the ESIPT process in PUBO. But different from the classical ESIPT-inhibition mechanism, the ESIPT process in the sensor PUBO is inhibited by the high energy barrier of its deprotonated form rather than by the absence of the transferred proton.
Co-reporter:Runze Liu, Yinghe Zhao and Tianshu Chu
Chemical Communications 2015 - vol. 51(Issue 12) pp:NaN2432-2432
Publication Date(Web):2015/01/01
DOI:10.1039/C4CC09424G
We studied the reaction mechanism of di-n-butylmagnesium decomposing into MgH2 in cyclohexane, and found a new route easier than famous β-hydride elimination. Further, we explored the dynamic behavior of graphene nano-flakes and MgH2 in cyclohexane, and gained new insights for efficient hydrogen storage material preparation.




![Benzamide, N-[3-(2-benzothiazolyl)-4-hydroxyphenyl]-](/data/chemimg/3119900/2533-14-4.png)
![Benzamide, N-[3-(2-benzothiazolyl)-4-hydroxyphenyl]-](/data/chemimg/3119900/2533-14-4_b.png)

