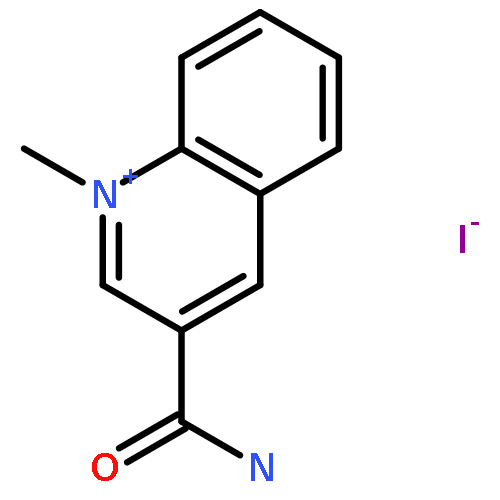
Co-reporter:Lu Wang, Joji Yui, Qifan Wang, Yiding Zhang, Wakana Mori, Yoko Shimoda, Masayuki Fujinaga, Katsushi Kumata, Tomoteru Yamasaki, Akiko Hatori, Benjamin H. Rotstein, Thomas Lee Collier, Chongzhao Ran, Neil Vasdev, Ming-Rong Zhang, and Steven H. Liang
ACS Chemical Neuroscience 2016 Volume 7(Issue 1) pp:109
Publication Date(Web):October 27, 2015
DOI:10.1021/acschemneuro.5b00248
Fatty acid amide hydrolase (FAAH) is one of the principle enzymes for metabolizing endogenous cannabinoid neurotransmitters such as anandamide, and thus regulates endocannabinoid (eCB) signaling. Selective pharmacological blockade of FAAH has emerged as a potential therapy to discern the endogenous functions of anandamide-mediated eCB pathways in anxiety, pain, and addiction. Quantification of FAAH in the living brain by positron emission tomography (PET) would help our understanding of the endocannabinoid system in these conditions. While most FAAH radiotracers operate by an irreversible (“suicide”) binding mechanism, a FAAH tracer with reversibility would facilitate quantitative analysis. We have identified and radiolabeled a reversible FAAH inhibitor, 7-(2-[11C]methoxyphenyl)-1-(5-(pyridin-2-yl)oxazol-2-yl)heptan-1-one ([11C]MPPO) in 13% radiochemical yield (nondecay corrected) with >99% radiochemical purity and 2 Ci/μmol (74 GBq/μmol) specific activity. The tracer showed moderate brain uptake (0.8 SUV) with heterogeneous brain distribution. However, blocking studies with a potent FAAH inhibitor URB597 demonstrated a low to modest specificity to the target. Measurement of lipophilicity, metabolite, and efflux pathway analysis were also performed to study the pharmacokinetic profile of [11C]MPPO. In all, we reported an efficient radiolabeling and preliminary evaluation of the first-in-class FAAH inhibitor [11C]MPPO with α-ketoheterocyclic scaffold.Keywords: FAAH; fatty acid amide hydrolase; PET; radiotracer; [11C]MPPO
Co-reporter:Yoko Shimoda, Masayuki Fujinaga, Akiko Hatori, Joji Yui, Yiding Zhang, Nobuki Nengaki, Yusuke Kurihara, Tomoteru Yamasaki, Lin Xie, Katsushi Kumata, Hideki Ishii, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 4) pp:627-634
Publication Date(Web):15 February 2016
DOI:10.1016/j.bmc.2015.12.026
To visualize fatty acid amide hydrolase (FAAH) in brain in vivo, we developed a novel positron emission tomography (PET) ligand N-(3,4-dimethylisoxazol-5-yl)piperazine-4-[4-(2-fluoro-4-[11C]methylphenyl)thiazol-2-yl]-1-carboxamide ([11C]DFMC, [11C]1). DFMC (1) was shown to have high binding affinity (IC50: 6.1 nM) for FAAH. [11C]1 was synthesized by C–11C coupling reaction of arylboronic ester 2 with [11C]methyl iodide in the presence of Pd catalyst. At the end of synthesis, [11C]1 was obtained with a radiochemical yield of 20 ± 10% (based on [11C]CO2, decay-corrected, n = 5) and specific activity of 48–166 GBq/μmol. After the injection of [11C]1 in mice, high uptake of radioactivity (>2% ID/g) was distributed in the lung, liver, kidney, and brain, organs with high FAAH expression. PET images of rat brains for [11C]1 revealed high uptakes in the cerebellar nucleus (SUV = 2.4) and frontal cortex (SUV = 2.0), two known brain regions with high FAAH expression. Pretreatment with the FAAH-selective inhibitor URB597 reduced the brain uptake. Higher than 90% of the total radioactivity in the rat brain was irreversible at 30 min after the radioligand injection. The present results indicate that [11C]1 is a promising PET ligand for imaging of FAAH in living brain.
Co-reporter:Masayuki Fujinaga, Tomoteru Yamasaki, Nobuki Nengaki, Masanao Ogawa, Katsushi Kumata, Yoko Shimoda, Joji Yui, Lin Xie, Yiding Zhang, Kazunori Kawamura, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 2) pp:370-374
Publication Date(Web):15 January 2016
DOI:10.1016/j.bmcl.2015.12.008
ADX88178 (1) has been recently developed as a potent positive allosteric modulator for metabotropic glutamate receptor 4 (mGluR4). The aim of this study was to develop [11C]1 as a novel positron emission tomography ligand and to evaluate its binding ability for mGluR4. Using stannyl precursor 3, [11C]1 was efficiently synthesized by introducing an [11C]methyl group into a pyrimidine ring via C–11C coupling and deprotection reactions, in 16 ± 6% radiochemical yield (n = 10). At the end of synthesis, 0.54–1.10 GBq of [11C]1 was acquired with >98% radiochemical purity and 90–120 GBq/μmol of specific activity. In vitro autoradiography and ex vivo biodistribution study in rat brains showed specific binding of [11C]1 in the cerebellum, striatum, thalamus, cerebral cortex, and medulla oblongata, which showed dose-dependent decreases by administration with multi-dose of unlabeled 1.
Co-reporter:Masayuki Fujinaga; Lin Xie; Tomoteru Yamasaki; Joji Yui; Yoko Shimoda; Akiko Hatori; Katsushi Kumata; Yiding Zhang; Nobuki Nengaki; Kazunori Kawamura
Journal of Medicinal Chemistry 2015 Volume 58(Issue 3) pp:1513-1523
Publication Date(Web):January 20, 2015
DOI:10.1021/jm501845n
Metabotropic glutamate 1 (mGlu1) receptor is found not only in the brain but also in melanomas and breast cancers. mGlu1 is a promising target for molecular imaging-based diagnosis and treatment of melanoma because its overexpression induces melanocyte carcinogenesis. Here we developed three PET tracers: 4-halogeno-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol- 2-yl]-N-[11C]methylbenzamide ([11C]4–6), which exhibited high uptake in target tumor and decreased uptake in nontarget brain tissues. In vitro binding assay indicated high to moderate binding affinities of 4–6 (Ki, 22–143 nM) for mGlu1 receptor. In vivo biodistribution studies in mice implanted with B16F10 melanoma cells confirmed high radioactive uptake in tumor and low uptake in blood, skin, and muscles. Inhibition of mGlu1 receptor using an mGlu1-selective ligand led to reduced radioactive uptake in the tumor. [11C]6 displayed the highest ratio of uptake between tumor and nontarget tissue and may prove useful as a PET tracer for mGlu1 imaging in melanoma.
Co-reporter:Katsushi Kumata, Joji Yui, Akiko Hatori, Jun Maeda, Lin Xie, Masanao Ogawa, Tomoteru Yamasaki, Yuji Nagai, Yoko Shimoda, Masayuki Fujinaga, Kazunori Kawamura, and Ming-Rong Zhang
ACS Chemical Neuroscience 2015 Volume 6(Issue 2) pp:339
Publication Date(Web):November 14, 2014
DOI:10.1021/cn500269g
We developed 2-methylpyridin-3-yl-4-(5-(2-fluorophenyl)-4H-1,2,4-triazol-3-yl)piperidine-1-[11C]carboxylate ([11C]MFTC) as a promising PET tracer for in vivo imaging of fatty acid amide hydrolase (FAAH) in rat and monkey brains. [11C]MFTC was synthesized by reacting 3-hydroxy-2-methylpyridine (2) with [11C]phosgene ([11C]COCl2), followed by reacting with 4-(5-(2-fluorophenyl)-4H-1,2,4-triazol-3-yl)piperidine (3), with a 20 ± 4.6% radiochemical yield (decay-corrected, n = 30) based on [11C]CO2 and 40 min synthesis time from the end of bombardment. A biodistribution study in mice showed high uptake of radioactivity in FAAH-rich organs, including the lung, liver, and kidneys. Positron emission tomography (PET) summation images of rat brains showed high radioactivity in the frontal cortex, cerebellum, and hippocampus, which was consistent with the regional distribution pattern of FAAH in rodent brain. Pretreatment with MFTC or FAAH-selective URB597 significantly reduced the uptake in the brain. PET imaging of monkey brain showed relatively high uptake in the whole brain, particularly in the occipital cortex, which was also inhibited by treatment with MFTC or URB597. More than 96% of the total radioactivity was irreversible in the brain homogenate of rats 5 min after the radiotracer injection. The specific in vivo FAAH binding indicates that [11C]MFTC is a promising PET tracer for visualizing FAAH in the brain.Keywords: FAAH; fatty acid amide hydrolase; irreversible specific binding; PET; [11C]COCl2; [11C]MFTC
Co-reporter:A. K. Tiwari, J. Yui, Pooja, S. Aggarwal, T. Yamasaki, L. Xie, N. Chadha, Y. Zhang, M. Fujinaga, Y. Shimoda, K. Kumata, A. K. Mishra, M. Ogawa and M.-R. Zhang
RSC Advances 2015 vol. 5(Issue 25) pp:19752-19759
Publication Date(Web):10 Feb 2015
DOI:10.1039/C4RA15833D
The 5-HT7 receptor is a recently cloned G-protein-coupled receptor (GPCR) that is important in regulating sleep, depression, and circadian rhythms. However, the potential pathophysiological roles of 5-HT7 have not been fully elucidated, and no 5-HT7 positron emission tomography (PET) radioligands are available, thus limiting imaging studies of this receptor in humans. Here, we present the radiosynthesis and biological evaluation of 5-(4-([11C]methoxyphenyl)-1-methyl-4-nitro-1H-imidazole ([11C]1) as a new PET ligand for 5-HT7. Three-dimensional pharmacophore evaluation and docking studies confirmed its high affinity for 5-HT7, and in vitro binding assays showed that the binding affinity was 16.8 ± 0.9 nM. The specific activity was found to be 48 ± 29 GBq μmol−1 for [11C]1 in a synthetic time of 26 ± 3 min (n = 8), having 38 ± 7% radiochemical yield (decay-corrected) based on [11C]CO2. Ligand interactions with human serum albumin were studied by fluorescence quenching to obtain a Stern–Volmer plot, which showed a binding constant of 1.15 × 104 M−1. Whole-body biodistribution patterns were evaluated in normal mice by 1 h dynamic PET imaging; this analysis showed rapid clearance of radioactivity from the main peripheral organs, with the exception of the liver. Preliminary PET studies in rat brains showed rapid accumulation of radioactivity in the brain. The regional radioactivity reached a maximum within 0–2 min after the radioligand injection and then decreased rapidly, resulting in minimal radiation burden in the brain during the scan. In summary, this specific biaryl system has shown potential as a 5-HT7 ligand and further optimization and longitudinal studies may yield the first small molecule-based PET ligand for 5-HT7 in clinical settings.
Co-reporter:Anjani K. Tiwari, Joji Yui, Yiding Zhang, Masayuki Fujinaga, Tomoteru Yamasaki, Lin Xie, Yoko Shimoda, Katsushi Kumata, Akiko Hatori and Ming-Rong Zhang
RSC Advances 2015 vol. 5(Issue 123) pp:101447-101454
Publication Date(Web):13 Nov 2015
DOI:10.1039/C5RA22594A
The five transmembrane translocator protein (18 kDa, TSPO) is abundantly expressed in the mitochondria of activated microglia (brain) and peripheral tissues, including those of the heart, lung and kidney. We recently developed the 18F-labelled molecule 2-[5-(4-[18F]fluoropropyloxyphenyl)-2-oxo-1,3-benzoxazol-3(2H)-yl]-N-methyl-N-phenylacetamide ([18F]FPBMP) as a novel positron emission tomography (PET) radioligand for imaging TSPO. In this study, we have evaluated the pharmacokinetics of this radioligand based on its biodistribution in mice, as well as the results of PET and metabolite studies in rats. The specificity of [18F]FPBMP towards TSPO was assessed by blocking experiments involving the intravenous injection of 1 mg kg−1 of unlabeled PK11195. A metabolite study was performed in the plasma and peripheral organs of rats by HPLC methods. The ex vivo biodistribution of [18F]FPBMP in mice showed a high uptake of radioactivity in TSPO enriched peripheral organs, especially in the lung, heart and kidney. The in vivo biodistribution of this compound was evaluated through PET summation images of rats 1–10, 10–20, 20–30 and 50–60 min after the injection of the radioligand. The TSPO-enriched organs, including the heart, kidney and lung, were clearly visualized. Pre-treatment with TSPO-specific PK11195 minimized the uptake of [18F]FPBMP in the TSPO-enriched tissues, thereby confirming its selectivity for TSPO. Metabolite analysis in rats confirmed the presence of [18F]FPBMP in the heart, lung and kidney up to 60 min. In summary, these data demonstrate that [18F]FPBMP is a suitable PET ligand for the imaging of TSPO in peripheral tissues.
Co-reporter:Y. Shimoda, J. Yui, Y. Zhang, A. Hatori, M. Ogawa, M. Fujinaga, T. Yamasaki, L. Xie, K. Kumata and M.-R. Zhang
RSC Advances 2015 vol. 5(Issue 128) pp:106122-106127
Publication Date(Web):03 Dec 2015
DOI:10.1039/C5RA22500K
We developed a novel positron emission tomography (PET) radiotracer N-(3,4-dimethylisoxazol-5-yl)piperazine-4-[4-(4-fluorophenyl)thiazol-2-yl]-1-[11C]carboxamide ([11C]DPFC, [11C]1) for in vivo imaging of fatty acid amide hydrolase (FAAH) in rat brain. Compound 1 showed a high binding affinity for FAAH (IC50: 3.3 nM). [11C]1 was synthesized by reaction of 5-amino-3,4-dimethylisoxazole (2) with [11C]phosgene ([11C]COCl2), followed by reaction with 4-(4-fluorophenyl)-2-(piperazin-1-yl)thiazole (3), with a 9 ± 4% radiochemical yield (decay-corrected, n = 9) based on [11C]CO2. A biodistribution study in mice showed a high uptake of radioactivity in FAAH-rich organs, including the lung, liver, and kidney. PET summation images of rat brains showed high radioactivity (>2 SUV) in the cerebellar nuclei and frontal cortex. This pattern was consistent with the known regional distribution pattern of FAAH in the rodent brain. Pretreatment with the FAAH-selective inhibitor URB597 significantly reduced the whole brain uptake of [11C]1. At 30 min after the radiotracer injection, more than 95% of the total radioactivity was found to be irreversible in the brain homogenate of rats. Our results indicate that [11C]1 is a promising PET tracer for in vivo visualization of FAAH in living brains.
Co-reporter:Katsushi Kumata, Joji Yui, Lin Xie, Yiding Zhang, Nobuki Nengaki, Masayuki Fujinaga, Tomoteru Yamasaki, Yoko Shimoda, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 16) pp:3230-3233
Publication Date(Web):15 August 2015
DOI:10.1016/j.bmcl.2015.05.085
Three compounds 1–3 containing methyl-sufanyl, sufinyl, or sulfonyl groups are strong inhibitors of glycogen synthase kinase 3β (GSK-3β), an enzyme associated with Alzheimer’s disease. We labeled 1–3 with 11C for a positron emission tomography (PET) brain imaging study. A novel thiophenol precursor 4 for radiosynthesis was prepared by reacting sulfoxide 2 with trifluoroacetic anhydride. [11C]1 was synthesized by reacting 4 with [11C]methyl iodide in 52 ± 5% radiochemical yield (n = 5, based on [11C]CO2, corrected for decay). Oxidation of [11C]1 with Oxone® produced [11C]2 and [11C]3, respectively. PET with [11C]1 and [11C]3 showed 2 fold higher brain uptake of radioactivity in a mouse model of cold water stress in which GSK-3β expression was increased, than in the controls.
Co-reporter:Anjani K. Tiwari, Masayuki Fujinaga, Joji Yui, Tomoteru Yamasaki, Lin Xie, Katsushi Kumata, Anil K. Mishra, Yoko Shimoda, Akiko Hatori, Bin Ji, Masanao Ogawa, Kazunori Kawamura, Feng Wang and Ming-Rong Zhang
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 47) pp:9621-9630
Publication Date(Web):01 Oct 2014
DOI:10.1039/C4OB01933D
The visualization of the activated microglia/TSPO is one of the main aspects of neuroimaging. Here we describe two new 18F-labelled molecules, 2-[5-(4-[18F]fluoroethoxyphenyl)- ([18F]2) and 2-[5-(4-[18F]fluoropropyloxyphenyl)- ([18F]3) -2-oxo-1,3-benzoxazol-3(2H)-yl]-N-methyl-N-phenylacetamide as novel PET ligands for imaging the translocator protein (18 kDa, TSPO) in the brain. The three-D pharmacophore evaluation and docking studies suggested their high affinity for the TSPO and in vitro binding assays of the TSPO showed binding affinities 6.6 ± 0.7 nM and 16.7 ± 2.5 nM for 2 and 3, respectively. The radiochemical yields for [18F]2 and [18F]3 were found to be 22 ± 4% (n = 8) and 5 ± 2% (n = 5), respectively at EOB. The radiochemical purity for both was found ≥98% and the specific activity was in the range of 98–364 GBq μmol−1 at EOS. In vitro autoradiography with an ischemic rat brain showed significantly increased binding on the ipsilateral side compared to the contralateral side. The specificity of [18F]2 and [18F]3 for binding TSPO was confirmed using the TSPO ligands PK11195 and MBMP. The biodistribution patterns of both PET ligands were evaluated in normal mice by 1 h dynamic PET imaging. In the brain, regional radioactivity reached the maximum very rapidly within 0–4 min for both ligands, similar to (R)[11C]PK11195. The metabolite study of [18F]2 also favoured a more favourable profile for quantification in comparison to (R)[11C]PK11195. In summary, these data indicated that [18F]2 and [18F]3 have good potential to work as PET ligands, therefore there are merits to use these radioligands for the in vivo evaluation in animal models to see their efficacy in the living brain.
Co-reporter:Anjani K. Tiwari;Joji Yui;Masayuki Fujinaga;Katsushi Kumata;Yoko Shimoda;Tomoteru Yamasaki;Lin Xie;Akiko Hatori;Jun Maeda;Nobuki Nengaki
Journal of Neurochemistry 2014 Volume 129( Issue 4) pp:712-720
Publication Date(Web):
DOI:10.1111/jnc.12670
Co-reporter:Yoko Shimoda, Joji Yui, Masayuki Fujinaga, Lin Xie, Katsushi Kumata, Masanao Ogawa, Tomoteru Yamasaki, Akiko Hatori, Kazunori Kawamura, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 15) pp:3574-3577
Publication Date(Web):1 August 2014
DOI:10.1016/j.bmcl.2014.05.045
CEP-32496 is a novel, orally active serine/threonine-protein kinase B-raf (BRAF) (V600E) kinase inhibitor that is being investigated in clinical trials for the treatment of some cancers in patients. In this study, we developed [11C-carbonyl]CEP-32496 as a novel positron emission tomography (PET) probe to study its biodistribution in the whole bodies of mice. [11C]CEP-32496 was synthesized by the reaction of 5-(1,1,1-trifluoro-2-methylpropan-2-yl)isoxazol-3-amine hydrochloride (1·HCl) with [11C]phosgene, followed by treatment with 3-(6,7-dimethoxyquinozolin-4-yloxy)aniline (2). Small-animal PET studies with [11C]CEP-32496 indicated that radioactivity levels (AUC0–90 min, SUV × min) accumulated in the brains of P-gp/BCRP knockout mice at a 8-fold higher rate than in the brains of wild-type mice.
Co-reporter:Yoko Shimoda, Joji Yui, Lin Xie, Masayuki Fujinaga, Tomoteru Yamasaki, Masanao Ogawa, Nobuki Nengaki, Katsushi Kumata, Akiko Hatori, Kazunori Kawamura, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 17) pp:5316-5322
Publication Date(Web):1 September 2013
DOI:10.1016/j.bmc.2013.06.020
1-[2-(4-Methoxyphenyl)phenyl]piperazine (4) is a potent serotonin 5-HT7 receptor antagonist (Ki = 2.6 nM) with a low binding affinity for the 5-HT1A receptor (Ki = 476 nM). As a potential positron emission tomography (PET) radiotracer for the 5-HT7 receptor, [11C]4 was synthesized at high radiochemical yield and specific activity, by O-[11C]methylation of 2′-(piperazin-1-yl)-[1,1′-biphenyl]-4-ol (6) with [11C]methyl iodide. Autoradiography revealed that [11C]4 showed in vitro specific binding with 5-HT7 in the rat brain regions, such as the thalamus which is a region with high 5-HT7 expression. Metabolite analysis indicated that intact [11C]4 in the brain exceeded 90% of the radioactive components at 15 min after the radiotracer injection, although two radiolabeled metabolites were found in the rat plasma. The PET study of rats showed moderated uptake of [11C]4 in the brain (1.2 SUV), but no significant regional difference in radioactivity in the brain. Pretreatment with 5-HT7-selective antagonist SB269970 (3) did not decrease the uptake of [11C]4 in the rat brain. Further studies are warranted that focus on the development of PET ligand candidates with higher binding affinity for 5-HT7 and higher in vivo stability in brain than 4.
Co-reporter:Masayuki Fujinaga ; Tomoteru Yamasaki ; Joji Yui ; Akiko Hatori ; Lin Xie ; Kazunori Kawamura ; Chiharu Asagawa ; Katsushi Kumata ; Yuichiro Yoshida ; Masanao Ogawa ; Nobuki Nengaki ; Toshimitsu Fukumura
Journal of Medicinal Chemistry 2012 Volume 55(Issue 5) pp:2342-2352
Publication Date(Web):February 8, 2012
DOI:10.1021/jm201590g
We designed three novel positron emission tomography ligands, N-(4-(6-(isopropylamino)pyrimidin-4-yl)-1,3-thiazol-2-yl)-4-[11C]methoxy-N-methylbenzamide ([11C]6), 4-[18F]fluoroethoxy-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide ([18F]7), and 4-[18F]fluoropropoxy-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide ([18F]8), for imaging metabotropic glutamate receptor type 1 (mGluR1) in rodent brain. Unlabeled compound 6 was synthesized by benzoylation of 4-pyrimidinyl-2-methylaminothiazole 10, followed by reaction with isopropylamine. Removal of the methyl group in 6 gave phenol precursor 12 for radiosynthesis. Two fluoroalkoxy analogues 7 and 8 were prepared by reacting 12 with tosylates 13 and 14. Radioligands [11C]6, [18F]7, and [18F]8 were synthesized by O-[11C]methylation or [18F]fluoroalkylation of 12. Compound 6 showed high in vitro binding affinity for mGluR1, whereas 7 and 8 had weak affinity. Autoradiography using rat brain sections showed that [11C]6 binding is aligned with the reported distribution of mGluR1 with high specific binding in the cerebellum and thalamus. PET study with [11C]6 in rats showed high brain uptake and a similar distribution pattern to that in autoradiography, indicating the usefulness of [11C]6 for imaging brain mGluR1.
Co-reporter:Masayuki Fujinaga ; Tomoteru Yamasaki ; Jun Maeda ; Joji Yui ; Lin Xie ; Yuji Nagai ; Nobuki Nengaki ; Akiko Hatori ; Katsushi Kumata ; Kazunori Kawamura
Journal of Medicinal Chemistry 2012 Volume 55(Issue 24) pp:11042-11051
Publication Date(Web):November 29, 2012
DOI:10.1021/jm301597s
Three novel 4-substituted benzamides have been synthesized as potential ligands for the positron emission tomography (PET) imaging of metabotropic glutamate 1 (mGlu1) receptor in the brain. Of these compounds, N-(4-(6-(isopropylamino)pyrimidin-4-yl)-1,3-thiazol-2-yl)-N,4-dimethylbenzamide (4) exhibited the highest binding affinity (Ki = 13.6 nM) for mGlu1 and was subsequently labeled with carbon-11. In vitro autoradiography using rat brain sections showed that [11C]4 binding was consistent with the distribution of mGlu1, with high specific binding in the cerebellum and thalamus. PET studies with [11C]4 in monkey showed a high brain uptake and a kinetic profile suitable for quantitative analysis. Pretreatment with a mGlu1-selective ligand 16 largely decreased the brain uptake, indicating high in vivo specific binding of [11C]4 to mGlu1. In metabolite analysis, only unchanged [11C]4 was found in the brain. [11C]4 is a useful PET ligand for the imaging and quantitative analysis of mGlu1 in monkey brain and merits further evaluation in humans.
Co-reporter:Masayuki Fujinaga;Jun Maeda;Joji Yui;Akiko Hatori;Tomoteru Yamasaki;Kazunori Kawamura;Katsushi Kumata;Yuichiro Yoshida;Yuji Nagai;Makoto Higuchi;Tetsuya Suhara;Toshimitsu Fukumura
Journal of Neurochemistry 2012 Volume 121( Issue 1) pp:115-124
Publication Date(Web):
DOI:10.1111/j.1471-4159.2011.07348.x
J. Neurochem. (2012) 121, 115–124.
Abstract
We developed 1-(2-[18F]fluoro-3-pyridyl)-4-(2-isopropyl-1-oxo-isoindoline-5-yl)-5-methyl-1H-1,2,3-triazole ([18F]FPIT) as a promising positron emission tomography (PET) ligand for in vitro and in vivo imaging of metabotropic glutamate receptor type 1 (mGluR1) in rat and monkey brains. In vitro autoradiography with [18F]FPIT was used to determine the distribution of radioactivity in rat and monkey brains. In vivo experiments were performed using dissection and small-animal PET on rats, and PET on monkey. Metabolite analysis was performed on rat plasma and brain, and monkey plasma. Autoradiography of rat and monkey brains showed that [18F]FPIT binding is aligned with the reported distribution of mGluR1 with high specific binding in the cerebellum and thalamus. PET study on rat and monkey showed high brain uptake and distribution patterns consistent with those seen in the autoradiographic studies. The radioactivity in the brain was significantly decreased by pre-treatment with unlabeled FPIT, indicative of a specific signal for mGluR1 that was inhibited by mGluR1-selective ligand JNJ-16259865 in the brain. Metabolite analysis showed that the percentage of unchanged [18F]FPIT was 89% in the brain homogenate of rat at 90 min after injection. In the monkey plasma, the percentage of unchanged form was 50% at 90 min. [18F]FPIT produced in vitro and in vivo signals to visualize mGluR1 expression in rat and monkey brains, suggesting the usefulness of [18F]FPIT for imaging mGluR1 in human brain.
Co-reporter:Katsushi Kumata, Masanao Ogawa, Makoto Takei, Masayuki Fujinaga, Yuichiro Yoshida, Nobuki Nengaki, Toshimitsu Fukumura, Kazutoshi Suzuki, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 1) pp:305-310
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmc.2011.10.077
Dantrolene (1) is a substrate for breast cancer resistant protein, which is widely distributed in the blood–brain-barrier, intestine, gall bladder, and liver. PET study with 1 labeled with a positron emitter can be used to visualize BCRP and to elucidate the effect of BCRP on the pharmacokinetics of drugs. The objective of this study was to label 1 using nitrogen-13 (13N, a positron emitter; half-life: 9.9 min). Using no-carrier-added [13N]NH3 as the labeling agent, we synthesized [13N]dantrolene ([13N]1) for the first time. The reaction of carbomyl chloride 2b with [13N]NH3 gave an unsymmetrical urea [13N]3, followed by cyclization of [13N]3 to afford [13N]1. Due to its instability, 2b was prepared in situ by treating amine 5 with triphosgene in a ratio of 4 to 1 and used for subsequent [13N]ammonolysis without purification.
Co-reporter:Chiharu Asakawa, Masanao Ogawa, Masayuki Fujinaga, Katsushi Kumata, Lin Xie, Tomoteru Yamasaki, Joji Yui, Toshimitsu Fukumura, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 11) pp:3594-3597
Publication Date(Web):1 June 2012
DOI:10.1016/j.bmcl.2012.04.049
N-(2-{3-[3,5-Bis(trifluoromethyl)]phenylureido}ethyl)glycyrrhetinamide (2), an ureido-substituted derivative of glycyrrhetinic acid (1), has been reported to display potent inhibitory activity for proteasome and kinase, which are overexpressed in tumors. In this study, we labeled this unsymmetrical urea 2 using [11C]phosgene ([11C]COCl2) as a labeling agent with the expectation that [11C]2 could become a positron emission tomography ligand for the imaging of proteasome and kinase in tumors. The strategy for the radiosynthesis of [11C]2 was to react hydrochloride of 3,5-bis(trifluoromethyl)aniline (4·HCl) with [11C]COCl2 to possibly give isocyanate [11C]6, followed by the reaction of [11C]6 with N-(2-aminoethyl)glycyrrhetinamide (3).
Co-reporter:Lin Xie;Tomoteru Yamasaki;Naotsugu Ichimaru
Journal of Neuroimmune Pharmacology 2012 Volume 7( Issue 1) pp:231-242
Publication Date(Web):2012 March
DOI:10.1007/s11481-011-9322-3
Neuroimaging measures have potential for monitoring neuroinflammation to guide treatment before the occurrence of significant functional impairment or irreversible neuronal damage in multiple sclerosis (MS). N-Benzyl-N-methyl-2-(7-[11C]methyl-8-oxo-2-phenyl-7,8-dihydro-9H-purin-9-yl) acetamide ([11C]DAC), a new developed positron emission tomography (PET) probe for translocator protein 18 kDa (TSPO), has been adopted to evaluate the neuroinflammation and treatment effects of experimental autoimmune encephalomyelitis (EAE), an animal model of MS. [11C]DAC-PET enabled visualization of neuroinflammation lesion of EAE by tracing TSPO expression in the spinal cords; the maximal uptake value reached in day 11 and 20 EAE rats with profound inflammatory cell infiltration compared with control, day 0 and 60 EAE rats. Biodistribution studies and in vitro autoradiography confirmed these in vivo imaging results. Doubling immunohistochemical studies showed the infiltration and expansion of CD4+ T cells and CD11b+ microglia; CD68+ macrophages were responsible for the increased TSPO levels visualized by [11C]DAC-PET. Furthermore, mRNA level analysis of the cytokines by quantitative reverse-transcription polymerase chain reaction (qRT-PCR) revealed that TSPO+/CD4 T cells, TSPO+ microglia and TSPO+ macrophages in EAE spinal cords were activated and secreted multiple proinflammation cytokines to mediate inflammation lesions of EAE. EAE rats treated with an immunosuppressive agent: 2-amino-2-[2-(4-octylphenyl)ethyl] propane-1,3-diolhydrochloride (FTY720), which exhibited an absence of inflammatory cell infiltrates, displaying a faint radioactive signal compared with the high accumulation of untreated EAE rats. These results indicated that [11C] DAC-PET imaging is a sensitive tool for noninvasively monitoring the neuroinflammation response and evaluating therapeutic interventions in EAE.
Co-reporter:Kazunori Kawamura;Masayuki Fujinaga;Joji Yui;Tetsuya Suhara;Yuichiro Yoshida;Jun Maeda;Toshimitsu Fukumura;Ming-Rong Zhang;Masaki Tokunaga;Makoto Higuchi;Yuji Nagai;Akiko Hatori;Tomoteru Yamasaki
European Journal of Nuclear Medicine and Molecular Imaging 2012 Volume 39( Issue 4) pp:632-641
Publication Date(Web):2012/04/01
DOI:10.1007/s00259-011-1995-6
In this study, we evaluate the utility of 4-[18F]fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide ([18F]FITM) as a positron emission tomography (PET) ligand for imaging of the metabotropic glutamate receptor subtype 1 (mGluR1) in rat and monkey brains.In vivo distribution of [18F]FITM in brains was evaluated by PET scans with or without the mGluR1-selective antagonist (JNJ16259685). Kinetic parameters of monkey PET data were obtained using the two-tissue compartment model with arterial blood sampling.In PET studies in rat and monkey brains, the highest uptake of radioactivity was in the cerebellum, followed by moderate uptake in the thalamus, hippocampus and striatum. The lowest uptake of radioactivity was detected in the pons. These uptakes in all brain regions were dramatically decreased by pre-administration of JNJ16259685. In kinetic analysis of monkey PET, the highest volume of distribution (VT) was detected in the cerebellum (VT = 11.5).[18F]FITM has an excellent profile as a PET ligand for mGluR1 imaging. PET with [18F]FITM may prove useful for determining the regional distribution and density of mGluR1 and the mGluR1 occupancy of drugs in human brains.
Co-reporter:Katsushi Kumata ; Joji Yui ; Akiko Hatori ; Masayuki Fujinaga ; Kazuhiko Yanamoto ; Tomoteru Yamasaki ; Kazunori Kawamura ; Hidekatsu Wakizaka ; Nobuki Nengaki ; Yuichiro Yoshida ; Masanao Ogawa ; Toshimitsu Fukumura
Journal of Medicinal Chemistry 2011 Volume 54(Issue 17) pp:6040-6049
Publication Date(Web):July 26, 2011
DOI:10.1021/jm200516a
To develop a PET ligand for imaging TSPO in peripheral organs, we designed three novel oxopurine analogues [11C]3a–c (LogD: 1.81–2.17) by introducing a pyridine ring in place of a benzene ring in the lead compound [11C]2 (LogD: 3.48). The desmethyl precursors 10 for radiosynthesis were synthesized by reacting glycine 7 with picolylamines 4, followed by hydrolysis and by Curtius rearrangement with diphenylphosphoryl azide. Methylation of 10a–c with methyl iodide produced unlabeled compounds 3a–c. The radiosynthesis of [11C]3a–c was performed by reacting 10a–c with [11C]methyl iodide. Compounds 3a–c displayed high or moderate in vitro binding affinities (Ki: 5–40 nM) for TSPO. PET with [11C]3a–c in rats showed high uptake in the lung, heart, and kidney, which are organs with high TSPO expression. Metabolite analysis with [11C]3a showed that radioactivity in these organs mainly corresponded with unchanged [11C]3a. PET with [11C]3a using a rat model of lung inflammation showed a significant signal in the lipopolysaccharide-treated lung.
Co-reporter:Masayuki Fujinaga, Tomoteru Yamasaki, Kazunori Kawamura, Katsushi Kumata, Akiko Hatori, Joji Yui, Kazuhiko Yanamoto, Yuichiro Yoshida, Masanao Ogawa, Nobuki Nengaki, Jun Maeda, Toshimitsu Fukumura, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 1) pp:102-110
Publication Date(Web):1 January 2011
DOI:10.1016/j.bmc.2010.11.048
The purpose of this study was to synthesize 6-[1-(2-[18F]fluoro-3-pyridyl)-5-methyl-1H-1,2,3-triazol-4-yl]quinoline ([18F]FPTQ, [18F]7a) and to evaluate its potential as a positron emission tomography ligand for imaging metabotropic glutamate receptor type 1 (mGluR1) in the rat brain. Compound [18F]7a was synthesized by [18F]fluorination of 6-[1-(2-bromo-3-pyridyl)-5-methyl-1H-1,2,3-triazol-4-yl]quinoline (7b) with potassium [18F]fluoride. At the end of synthesis, 1280–1830 MBq (n = 8) of [18F]7a was obtained with >98% radiochemical purity and 118–237 GBq/μmol specific activity using 3300–4000 MBq of [18F]F−. In vitro autoradiography showed that [18F]7a had high specific binding with mGluR1 in the rat brain. Biodistribution study using a dissection method and small-animal PET showed that [18F]7a had high uptake in the rat brain. The uptake of radioactivity in the cerebellum was reduced by unlabeled 7a and mGluR1-selective ligand JNJ-16259685 (2), indicating that [18F]7a had in vivo specific binding with mGluR1. Because of a low amount of radiolabeled metabolite present in the brain, [18F]7a may have a limiting potential for the in vivo imaging of mGluR1 by PET.
Co-reporter:Tomoteru Yamasaki, Masayuki Fujinaga, Yuichiro Yoshida, Katsushi Kumata, Joji Yui, Kazunori Kawamura, Akiko Hatori, Toshimitsu Fukumura, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 10) pp:2998-3001
Publication Date(Web):15 May 2011
DOI:10.1016/j.bmcl.2011.03.046
The purpose of this study was to develop 4-[18F]fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide ([18F]FITM, [18F]4) as a new PET ligand for imaging metabotropic glutamate receptor subtype 1 (mGluR1). [18F]4 was synthesized by [18F]fluorination of a novel nitro precursor 3 with [18F]KF in the presence of Kryptofix 222. At the end of synthesis, 429–936 MBq (n = 8) of [18F]4 was obtained with >99% radiochemical purity and 204–559 GBq/μmol specific activity starting from 6.7 to 13.0 GBq of [18F]F−. The brain distribution of [18F]4 was determined by the in vitro and ex vivo autoradiography using rat brain sections. The in vitro and in vivo specific binding of [18F]4 to mGluR1 was detected in the cerebellum, thalamus, hippocampus, and striatum. These results suggest that [18F]4 is a promising PET ligand for the in vivo evaluation of mGluR1.
Co-reporter:Chiharu Asakawa, Masanao Ogawa, Katsushi Kumata, Masayuki Fujinaga, Koichi Kato, Tomoteru Yamasaki, Joji Yui, Kazunori Kawamura, Akiko Hatori, Toshimitsu Fukumura, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 8) pp:2220-2223
Publication Date(Web):15 April 2011
DOI:10.1016/j.bmcl.2011.03.002
Sorafenib (Nexavar, BAY43-9006, 1) is a second-generation, orally active multikinase inhibitor that is approved for the treatment of some cancers in patients. In this Letter, we developed [11C]1 as a novel positron emission tomography (PET) probe, and evaluated the influence of ABC transporters-mediated efflux on brain uptake using PET with [11C]1 in P-glycoprotein (P-gp)/breast cancer resistance protein (Bcrp) knockout mice versus wild-type mice. [11C]1 was synthesized by the reaction of hydrochloride of aniline 2 with [11C]phosgene ([11C]COCl2) to give isocyanate [11C]6, followed by reaction with another aniline 3. Small-animal PET study with [11C]1 indicated that the radioactivity level (AUC0–60 min, SUV × min) in the brains of P-gp/Bcrp knockout mice was about three times higher than in wild-type mice.
Co-reporter:Chiharu Asakawa, Masanao Ogawa, Katsushi Kumata, Masayuki Fujinaga, Tomoteru Yamasaki, Lin Xie, Joji Yui, Kazunori kawamura, Toshimitsu Fukumura, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 23) pp:7017-7020
Publication Date(Web):1 December 2011
DOI:10.1016/j.bmcl.2011.09.102
Three ureido-substituted benzenesulfonamides 1a–c have been developed as potent inhibitors for carbonic anhydrase IX, which is overexpressed in hypoxic tumors. In this study, we labeled these unsymmetrical ureas 1a–c using [11C]phosgene ([11C]COCl2) as a labeling agent with the expectation that [11C]1a–c could become promising positron tomography probes for imaging carbonic anhydrase IX in tumors. The strategy for radiosynthesis of [11C]1a–c was to react hydrochloride of anilines 2a–c with [11C]COCl2 to give isocyanate [11C]4a–c, followed by a reaction with 4-aminobenzenesulfonamide (3).
Co-reporter:Masayuki Fujinaga, Katsushi Kumata, Kazuhiko Yanamoto, Kazunori Kawamura, Tomoteru Yamasaki, Joji Yui, Akiko Hatori, Masanao Ogawa, Yuichiro Yoshida, Nobuki Nengaki, Jun Maeda, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 5) pp:1565-1568
Publication Date(Web):1 March 2010
DOI:10.1016/j.bmcl.2010.01.074
Two novel triaryl ligands 2 and 5 with potent in vitro binding affinities for the cannabinoid subtype-2 (CB2) receptor were labeled with a positron-emitting radioactive nuclide 11C. Radioligands [11C]2, [11C]5, and their analogs [11C]3 and [11C]4 were synthesized by O-[11C]methylation of their corresponding phenol precursors with [11C]CH3I. [11C]2–5 had relatively high uptakes (>1.2% injected dose/g tissue) in mouse brains.
Co-reporter:Kazuhiko Yanamoto;Ming-Rong Zhang;Nobuhiko Takai;Katsushi Kumata;Akiko Hatori;Jun Toyohara;Kazunori Kawamura;Tomoteru Yamasaki;Sachiko Koike;Joji Yui;Kazutoshi Suzuki;Koichi Ando
Molecular Imaging and Biology 2010 Volume 12( Issue 2) pp:
Publication Date(Web):2010/04/01
DOI:10.1007/s11307-009-0265-5
Gefitinib (N-(3-chloro-4-fluorophenyl)-7-methoxy-6-[3-(morpholin-4-yl)propoxy]quinazolin-4-amine, Iressa) is an approved anticancer drug. In this study, we labeled gefitinib with carbon-11 and evaluated [11C]gefitinib to explore its specific binding in intact fibrosarcoma (NFSa)-bearing mice.[11C]Gefitinib was synthesized by the reaction of desmethyl precursor (1) with [11C]CH3I. In vitro uptake of [11C]gefitinib into NFSa, human-A431 epidermoid carcinoma, and Jurkat T cells was determined. Positron emission tomography (PET) imaging using [11C]gefitinib was performed for NFSa-bearing mice.[11C]Gefitinib accumulated into NFSa cells with 2.1 uptake ratio (UR)/mg protein in cells. Addition of nonradioactive gefitinib decreased uptake in a concentration-dependent manner. [11C]Gefitinib also had high uptake (2.6 UR/mg protein) into epidermal growth factor receptor/tyrosine kinase (EGFR/TK)-rich A431 cells but low uptake (0.2 UR/mg protein) into EGFR/TK-poor Jurkat cells. In vivo distribution study on NFSa-bearing mice by the dissection method revealed that [11C]gefitinib specifically accumulated into the tumor. The ratio of radioactivity in tumors to that in blood and muscle as two comparative regions increased from 0.4 to 6.0 and from 0.6 to 5.0 during this experiment (0–60 min), respectively. PET for NFSa-bearing mice produced a clear tumor image, although high radioactivity was distributed throughout the body. Treatment with nonradioactive gefitinib (100 mg/kg) decreased uptake in the tumor. In vivo metabolite analysis demonstrated that [11C]gefitinib was stable in the tumor, liver, kidney, and blood.These results demonstrated the promising potential of [11C]gefitinib to serve as a PET ligand for in vivo imaging of NFSa-bearing mice.
Co-reporter:Katsushi Kumata;Makoto Takei;Masanao Ogawa;Joji Yui;Akiko Hatori;Kazutoshi Suzuki
Journal of Labelled Compounds and Radiopharmaceuticals 2010 Volume 53( Issue 2) pp:53-57
Publication Date(Web):
DOI:10.1002/jlcr.1699
Abstract
Recent studies revealed that thalidomide (1) has unique and broad pharmacological effects on multi-targets although the application of 1 in therapy is still controversial. In this study, we synthesized nitrogen-13-labeled thalidomide ([13N]1) as a potential positron emission tomography (PET) probe using no-carrier-added [13N]NH3 as a labeling agent. By use of an automated system, [13N]1 was prepared by reacting N-phthaloylglutamic anhydride (2) with [13N]NH3, following by cyclization with carbonyldiimidazole in a radiochemical yield of 56±12% (based on [11N]NH3, corrected for decay) and specific activity of 49±24 GBq/µmol at the end of synthesis (EOS). At EOS, 570–780 MBq (n=7) of [13N]1 was obtained at a beam current of 15 µA after 15 min proton bombardment with a synthesis time of 14 min from the end of bombardment. Using a small animal PET scanner, preliminary biodistribution of [13N]1 in mice was examined. Copyright © 2010 John Wiley & Sons, Ltd.
Co-reporter:Kazuhiko Yanamoto, Katsushi Kumata, Tomoteru Yamasaki, Chika Odawara, Kazunori Kawamura, Joji Yui, Akiko Hatori, Kazutoshi Suzuki, Ming-Rong Zhang
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 6) pp:1707-1710
Publication Date(Web):15 March 2009
DOI:10.1016/j.bmcl.2009.01.093
[18F]FEAC ([18F]4a) and [18F]FEDAC ([18F]4b) were developed as two novel positron emission tomography (PET) ligands for peripheral-type benzodiazepine receptor (PBR). [18F]4a and [18F]4b were synthesized by fluoroethylation of precursors 8a and 8b with [18F]FCH2CH2Br ([18F]9), respectively. Small-animal PET scan for a neuroinflammatory rat model showed that the two radioligands had high uptakes of radioactivity in the kainic acid-infused striatum, a brain region where PBR density was increased.[18F]FEAC ([18F]4a) and [18F]FEDAC ([18F]4b), two potent PET ligands for peripheral-type benzodiazepine receptor, were synthesized and evaluated.
Co-reporter:Katsushi Kumata;Makoto Takei;Masanao Ogawa;Koichi Kato;Kazutoshi Suzuki
Journal of Labelled Compounds and Radiopharmaceuticals 2009 Volume 52( Issue 5) pp:166-172
Publication Date(Web):
DOI:10.1002/jlcr.1584
Abstract
The aim of this study was to develop a practical labeling method of [13N]ligands using no-carrier-added [13N]NH3 with high specific activity. [13N]urea analogues [13N]1a and [13N]2a or [13N]carbamate [13N]3a were synthesized by reacting isocyanate 5a, carbamoyl chloride 6a or chloroformate 7a with [13N]NH3. The precursors 5a–7a were prepared by treating amines 8a and 9a and alcohol 10a with triphosgene in situ. These reaction mixtures were not purified and were used directly for [13N]ammonolysis, respectively. Using the one-pot method, we synthesized [13N]carbamazepine ([13N]4), a putative positron emission tomography ligand for brain imaging. Copyright © 2009 John Wiley & Sons, Ltd.
Co-reporter:Fujiko Konno, Takuya Arai, Ming-Rong Zhang, Akiko Hatori, Kazuhiko Yanamoto, Masanao Ogawa, Gukuto Ito, Chika Odawara, Tomoteru Yamasaki, Koichi Kato, Kazutoshi Suzuki
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 4) pp:1260-1263
Publication Date(Web):15 February 2008
DOI:10.1016/j.bmcl.2008.01.037
Oseltamivir phosphate (Tamiflu®, 1·H3PO4 is an orally active anti-influenza drug, which is hydrolyzed by esterase to its carboxylate metabolite Ro 64-0802 (2) with potent activity inhibiting neuraminidase. In this study, for the first time, we synthesized carbon-11-labeled oseltamivir ([11C]1) and Ro 64-0802 ([11C]2) as two novel positron emission tomography probes and demonstrated that [11C]1 had twofold higher radioactivity concentration in the mouse brains than [11C]2.Radiosyntheses of [11C]oseltamivir and its active metabolite [11C]Ro 64-0802 as two positron emission tomography probes and their radioactivity concentrations in the mouse brains are reported for the first time.
Co-reporter:Ming-Rong Zhang, Jun Maeda, Takehito Ito, Takashi Okauchi, Masanao Ogawa, Junko Noguchi, Tetsuya Suhara, Christer Halldin, Kazutoshi Suzuki
Bioorganic & Medicinal Chemistry 2005 Volume 13(Issue 5) pp:1811-1818
Publication Date(Web):1 March 2005
DOI:10.1016/j.bmc.2004.11.058
N-(5-Fluoro-2-phenoxyphenyl)-N-(2-[18F]fluoromethoxy-d2-5-methoxybenzyl)acetamide ([18F]2) is a potent ligand (IC50: 1.71 nM) for peripheral benzodiazepine receptor (PBR). However, in vivo evaluation on rodents and primates showed that this ligand was unstable and rapidly metabolized to [18F]F− by defluorination of the [18F]fluoromethyl moiety. In this study, we designed a deuterium-substituted analogue, N-(5-fluoro-2-phenoxyphenyl)-N-(2-[18F]fluoromethoxy-d2-5-methoxybenzyl)acetamide ([18F]5) as a radioligand for PBR to reduce the in vivo metabolic rate of the non-deuterated [18F]2. The design principle was based on the hypothesis that the deuterium substitution may reduce the rate of defluorination initiated by cleavage of the C–H bond without altering the binding affinity for PBR. The non-radioactive 5 was prepared by reacting diiodomethane-d2 (CD2I2, 6) with a phenol precursor 7, followed by treatment with tetrabutylammonium fluoride. The ligand [18F]5 was synthesized by the alkylation of 7 with [18F]fluoromethyl iodide-d2 ([18F]FCD2I, [18F]9). Compound 5 displayed a similar in vitro affinity to PBR (IC50: 1.90 nM) with 2. In vivo evaluation demonstrated that [18F]5 was metabolized by defluorination to [18F]F− as a main radioactive component, but its metabolic rate was slower than that of [18F]2 in the brain of mice. The deuterium substitution decreased the radioactivity level of [18F]5 in the bone of mouse, augmented by the percentage of specific binding to PBR in the rat brain determined by ex vivo autoradiography. However, the PET image of [18F]5 for monkey brain showed high radioactivity in the brain and skull, suggesting a possible species difference between rodents and primates.
Co-reporter:Ming-Rong Zhang, Masanao Ogawa, Kenji Furutsuka, Yuichiro Yoshida, Kazutoshi Suzuki
Journal of Fluorine Chemistry 2004 Volume 125(Issue 12) pp:1879-1886
Publication Date(Web):December 2004
DOI:10.1016/j.jfluchem.2004.06.017
In this study, we report the synthesis and reactivity of [18F]fluoromethyl iodide ([18F]FCH2I) with various nucleophilic substrates and the stabilities of [18F]fluoromethylated compounds. [18F]FCH2I was prepared by reacting diiodomethane (CH2I2) with [18F]KF, and purified by distillation in radiochemical yields of 14–31% (n = 25). [18F]FCH2I was stable in organic solvents commonly used for labeling and aqueous solution with pH 1–7, but was unstable in basic solutions. [18F]FCH2I displayed a high reactivity with various nucleophilic substrates such as phenol, thiophenol, amide and amine. The [18F]fluoromethylated compounds synthesized by the reactions of phenol, thiophenol and tertiary amine with [18F]FCH2I were stable for purification, formulation and storage. In contrast, the [18F]fluoromethylated compounds synthesized by the reactions of primary or secondary amines, and amide with [18F]FCH2I were too unstable to be detected or purified from the reaction mixtures. Defluorination of these [18F]fluoromethyl compounds was a main decomposition route.[18F]Fluoromethyl iodide ([18F]FCH2I) which was prepared by reaction of diiodomethane with [18F]F− displayed high reactivities with phenol, thiophenol, amide and amine. Defluorination made some [18F]fluoromethylated products unstable.
Co-reporter:Ming-Rong Zhang, Kenji Furutsuka, Jun Maeda, Tatsuya Kikuchi, Takayo Kida, Takashi Okauchi, Toshiaki Irie, Kazutoshi Suzuki
Bioorganic & Medicinal Chemistry 2003 Volume 11(Issue 12) pp:2519-2527
Publication Date(Web):12 June 2003
DOI:10.1016/S0968-0896(03)00177-9
N-[18F]Fluoroethyl-4-piperidyl acetate ([18F]FEtP4A) was synthesized and evaluated as a PET tracer for imaging brain acetylcholinesterase (AchE) in vivo. [18F]FEtP4A was previously prepared by reacting 4-piperidyl acetate (P4A) with 2-[18F]fluoroethyl bromide ([18F]FEtBr) at 130 °C for 30 min in 37% radiochemical yield using an automated synthetic system. In this work, [18F]FEtP4A was synthesized by reacting P4A with 2-[18F]fluoroethyl iodide ([18F]FEtI) or 2-[18F]fluoroethyl triflate ([18F]FEtOTf in improved radiochemical yields, compared with [18F]FEtBr under the corresponding condition. Ex vivo autoradiogram of rat brain and PET summation image of monkey brain after iv injection of [18F]FEtP4A displayed a high radioactivity in the striatum, a region with the highest AchE activity in the brain. Moreover, the distribution pattern of 18F radioactivity was consistent with that of AchE in the brain: striatum>frontal cortex>cerebellum. In the rat and monkey plasma, two radioactive metabolites were detected. However, their presence might not preclude the imaging studies for AchE in the brain, because they were too hydrophilic to pass the blood–brain barrier and to enter the brain. In the rat brain, only [18F]fluoroethyl-4-piperidinol ([18F]FEtP4OH) was detected at 30 min postinjection. The hydrolytic [18F]FEtP4OH displayed a slow washout and a long retention in the monkey brain until the PET experiment (120 min). Although [18F]FEtP4A is a potential PET tracer for imaging AchE in vivo, its lower hydrolytic rate and lower specificity for AchE than those of [11C]MP4A may limit its usefulness for the quantitative measurement for AchE in the primate brain.[18F]FEtP4A, a potential PET tracer for imaging brain acetylcholinesterase (AchE) in vivo, was synthesized by the respective reaction of P4A with [18F]FEtBr, [18F]FEtBr/NaI and [18F]FEtOTf. After iv injection of [18F]FEtP4A into animals, ex vivo autoradiogram of rat brain and PET image of monkey brain displayed a significant radioactivity in the striatum, the region with the highest AchE activity in the brain.
Co-reporter:Ming-Rong Zhang, Jun Maeda, Kenji Furutsuka, Yuichiro Yoshida, Masanao Ogawa, Tetsuya Suhara, Kazutoshi Suzuki
Bioorganic & Medicinal Chemistry Letters 2003 Volume 13(Issue 2) pp:201-204
Publication Date(Web):20 January 2003
DOI:10.1016/S0960-894X(02)00886-7
We synthesized and evaluated N-(5-fluoro-2-phenoxyphenyl)-N-(2-[18F]fluoromethyl-5-methoxybenzyl)acetamide ([18F]-FMDAA1106) and N-(5-fluoro-2-phenoxyphenyl)-N-(2-[18F]fluoroethyl-5-methoxybenzyl)acetamide ([18F]FEDAA1106) as two potent radioligands for peripheral benzodiazepine receptors (PBR). [18F]FMDAA1106 and [18F]FEDAA1106 were respectively synthesized by fluoroalkylation of the desmethyl precursor DAA1123 with [18F]FCH2I and [18F]FCH2CH2Br. Ex vivo autoradiograms of [18F]FMDAA1106 and [18F]FEDAA1106 binding sites in the rat brains revealed that a high radioactivity was present in the olfactory bulb, the highest PBR density region in the brain.[18F]FMDAA1106 and [18F]FEDAA1106, two potent radioligands for peripheral benzodiazepine receptors (PBR), were designed and synthesized. Ex vivo autoradiograms of rat brains revealed that they had high specific bindings in the olfactory bulb, the region with the highest PBR density in the brain.
Co-reporter:Yuichiro Yoshida;Kenji Furutsuka;Kazutoshi Suzuki
Journal of Labelled Compounds and Radiopharmaceuticals 2003 Volume 46(Issue 6) pp:587-598
Publication Date(Web):4 APR 2003
DOI:10.1002/jlcr.703
[18F]Fluoroethyl bromide ([18F]FEtBr) is a useful synthetic precursor to synthesize 18F-labeled compounds. However, the lower reactivity of [18F]FEtBr with amine, phenol and amide functional groups than that of [11C]CH3I partly limits its wide application in the synthesis of [18F]fluoroethylated compounds. The aim of this study was to increase the reactivity of [18F]FEtBr with various nucleophilic substrates for PET tracers containing amine, phenol and amide moieties. The present strategies included (1) adding NaI into the reaction mixture of [18F]FEtBr and substrate, where [18F]FEtI is reversibly formed and becomes more reactive; (2) converting [18F]FEtBr into much more reactive [18F]FEtOTf, similar to conversion of [11C]CH3I into [11C]CH3OTf. By these efforts, the [18F]fluoroethylation efficiency of various substrates containing amine, phenol and amide groups with [18F]FEtBr/NaI and [18F]FEtOTf was significantly improved, compared with the corresponding reaction efficiency with [18F]FEtBr. Copyright © 2003 John Wiley & Sons, Ltd.
Co-reporter:Jun Maeda;Shigeru Obayashi;Terushi Haradahira;Kazutoshi Suzuki;Takashi Okauchi;Takayo Kida;Tetsuya Suhara
Journal of Labelled Compounds and Radiopharmaceuticals 2002 Volume 45(Issue 10) pp:857-866
Publication Date(Web):5 AUG 2002
DOI:10.1002/jlcr.606
DR4446 (1-methyl-2a-[4-(4,5,6,7-tetrahydrothieno[3,2-c]pyridin-5-yl)butyl]-2a,3,4,5-tetrahydro-1H-benz[cd]indole-2-one) is a potent 5-HT7 receptor antagonist (Ki=9.7 nM) with a high selectivity over other 5-HT family receptors (Ki for 5-HT1A: 770 nM; for other 5-HT receptors: >1000 nM). As a positron emission tomography (PET) tracer for the 5-HT7 receptor, [11C]DR4446 was synthesized at high radiochemical purity ( >98%) with specific activity of 73–120 GBq/μmol at the end of synthesis by the alkylation of the desmethyl precursor (1) with [11C]CH3I in the presence of NaH. A PET study in monkey demonstrated that [11C]DR4446 had good permeability into the brain, and had a specific binding component in the brain regions including the thalamus, possibly an area in the 5-HT7 receptors. Metabolite analysis showed that [11C]DR4446 was relatively stable and low percentages of two radio-labeled metabolites were detected in the plasma of monkey using HPLC. Copyright © 2002 John Wiley & Sons, Ltd.
Co-reporter:Ming-Rong Zhang, Katsushi Kumata, Makoto Takei, Toshimitsu Fukumura, Kazutoshi Suzuki
Applied Radiation and Isotopes (October 2008) Volume 66(Issue 10) pp:1341-1345
Publication Date(Web):October 2008
DOI:10.1016/j.apradiso.2008.04.011
Co-reporter:Takuya Arai, Ming-Rong Zhang, Masanao Ogawa, Toshimitsu Fukumura, Koichi Kato, Kazutoshi Suzuki
Applied Radiation and Isotopes (February 2009) Volume 67(Issue 2) pp:296-300
Publication Date(Web):February 2009
DOI:10.1016/j.apradiso.2008.09.013
Co-reporter:Anjani K. Tiwari, Masayuki Fujinaga, Joji Yui, Tomoteru Yamasaki, Lin Xie, Katsushi Kumata, Anil K. Mishra, Yoko Shimoda, Akiko Hatori, Bin Ji, Masanao Ogawa, Kazunori Kawamura, Feng Wang and Ming-Rong Zhang
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 47) pp:NaN9630-9630
Publication Date(Web):2014/10/01
DOI:10.1039/C4OB01933D
The visualization of the activated microglia/TSPO is one of the main aspects of neuroimaging. Here we describe two new 18F-labelled molecules, 2-[5-(4-[18F]fluoroethoxyphenyl)- ([18F]2) and 2-[5-(4-[18F]fluoropropyloxyphenyl)- ([18F]3) -2-oxo-1,3-benzoxazol-3(2H)-yl]-N-methyl-N-phenylacetamide as novel PET ligands for imaging the translocator protein (18 kDa, TSPO) in the brain. The three-D pharmacophore evaluation and docking studies suggested their high affinity for the TSPO and in vitro binding assays of the TSPO showed binding affinities 6.6 ± 0.7 nM and 16.7 ± 2.5 nM for 2 and 3, respectively. The radiochemical yields for [18F]2 and [18F]3 were found to be 22 ± 4% (n = 8) and 5 ± 2% (n = 5), respectively at EOB. The radiochemical purity for both was found ≥98% and the specific activity was in the range of 98–364 GBq μmol−1 at EOS. In vitro autoradiography with an ischemic rat brain showed significantly increased binding on the ipsilateral side compared to the contralateral side. The specificity of [18F]2 and [18F]3 for binding TSPO was confirmed using the TSPO ligands PK11195 and MBMP. The biodistribution patterns of both PET ligands were evaluated in normal mice by 1 h dynamic PET imaging. In the brain, regional radioactivity reached the maximum very rapidly within 0–4 min for both ligands, similar to (R)[11C]PK11195. The metabolite study of [18F]2 also favoured a more favourable profile for quantification in comparison to (R)[11C]PK11195. In summary, these data indicated that [18F]2 and [18F]3 have good potential to work as PET ligands, therefore there are merits to use these radioligands for the in vivo evaluation in animal models to see their efficacy in the living brain.

![3-[2-CYCLOHEXYL(HYDROXY)PHENYLACETOXY]-1-METHYL-1-AZONIABICYCLO[2.2.2]OCTANE BROMIDE](http://img.cochemist.com/ccimg/1005300/1005201-24-0.png)
![3-[2-CYCLOHEXYL(HYDROXY)PHENYLACETOXY]-1-METHYL-1-AZONIABICYCLO[2.2.2]OCTANE BROMIDE](http://img.cochemist.com/ccimg/1005300/1005201-24-0_b.png)
![1-Piperazinecarboxamide,N-(3,4-dimethyl-5-isoxazolyl)-4-[4-(4-fluorophenyl)-2-thiazolyl]-](http://img.cochemist.com/ccimg/887700/887623-12-3.png)
![1-Piperazinecarboxamide,N-(3,4-dimethyl-5-isoxazolyl)-4-[4-(4-fluorophenyl)-2-thiazolyl]-](http://img.cochemist.com/ccimg/887700/887623-12-3_b.png)
![2-PyriMidinaMine,4-Methyl-N-[5-Methyl-4-(1H-pyrazol-4-yl)-2-thiazolyl]-](http://img.cochemist.com/ccimg/1235400/1235318-89-4.png)
![2-PyriMidinaMine,4-Methyl-N-[5-Methyl-4-(1H-pyrazol-4-yl)-2-thiazolyl]-](http://img.cochemist.com/ccimg/1235400/1235318-89-4_b.png)
![Benzonitrile, 3-hydroxy-5-[5-(2-pyridinyl)-2H-tetrazol-2-yl]-](http://img.cochemist.com/ccimg/868200/868157-25-9.png)
![Benzonitrile, 3-hydroxy-5-[5-(2-pyridinyl)-2H-tetrazol-2-yl]-](http://img.cochemist.com/ccimg/868200/868157-25-9_b.png)
![BENZONITRILE, 3-METHYL-5-[5-(2-PYRIDINYL)-2H-TETRAZOL-2-YL]-](http://img.cochemist.com/ccimg/868200/868157-24-8.png)
![BENZONITRILE, 3-METHYL-5-[5-(2-PYRIDINYL)-2H-TETRAZOL-2-YL]-](http://img.cochemist.com/ccimg/868200/868157-24-8_b.png)
![2-(4-(Methylamino)phenyl)benzo[d]thiazol-6-ol](/data/chemimg/98400/566169-93-5.png)
![2-(4-(Methylamino)phenyl)benzo[d]thiazol-6-ol](/data/chemimg/98400/566169-93-5_b.png)


![Butanoic acid, 2-[(diphenylmethylene)amino]-, 1,1-dimethylethyl ester,(2R)-](http://img.cochemist.com/ccimg/219700/219601-71-5.png)
![Butanoic acid, 2-[(diphenylmethylene)amino]-, 1,1-dimethylethyl ester,(2R)-](http://img.cochemist.com/ccimg/219700/219601-71-5_b.png)














![Isoquinoline,5-[(2-methyl-1-piperazinyl)sulfonyl]-](http://img.cochemist.com/ccimg/84500/84477-87-2.png)
![Isoquinoline,5-[(2-methyl-1-piperazinyl)sulfonyl]-](http://img.cochemist.com/ccimg/84500/84477-87-2_b.png)
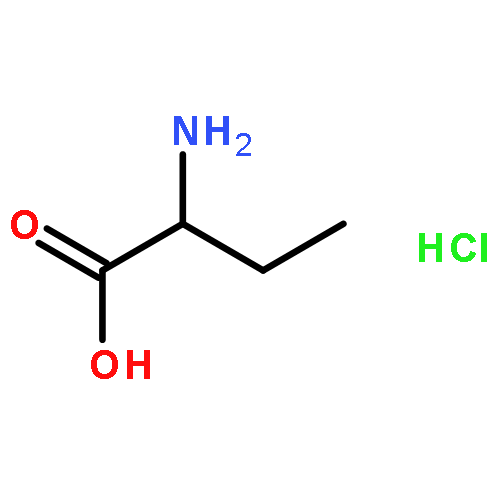
![ethyl [(3-methoxybenzoyl)amino]acetate](http://img.cochemist.com/ccimg/65700/65654-29-7.png)
![ethyl [(3-methoxybenzoyl)amino]acetate](http://img.cochemist.com/ccimg/65700/65654-29-7_b.png)












![AMMONIA; [(1R,2S,3R,4R,5S,6S)-2,3-DIHYDROXY-4,5,6-TRIPHOSPHONOOXY<WBR />-CYCLOHEXYL] DIHYDROGEN PHOSPHATE](http://img.cochemist.com/ccimg/7200/7114-81-0.png)
![AMMONIA; [(1R,2S,3R,4R,5S,6S)-2,3-DIHYDROXY-4,5,6-TRIPHOSPHONOOXY<WBR />-CYCLOHEXYL] DIHYDROGEN PHOSPHATE](http://img.cochemist.com/ccimg/7200/7114-81-0_b.png)
![1H-Imidazo[1,2-b]pyrazole,2,3-dihydro-](http://img.cochemist.com/ccimg/6800/6714-29-0.png)
![1H-Imidazo[1,2-b]pyrazole,2,3-dihydro-](http://img.cochemist.com/ccimg/6800/6714-29-0_b.png)
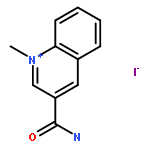








![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6_b.png)