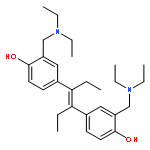
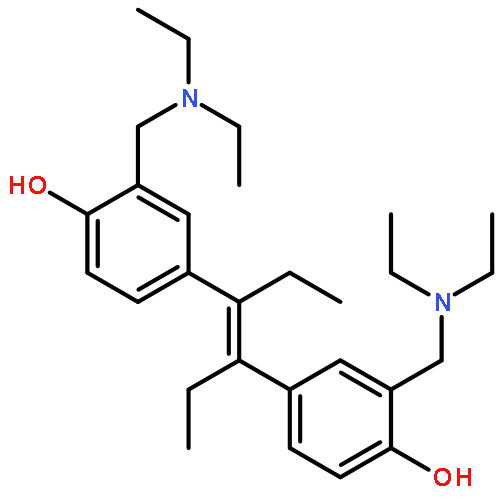
Co-reporter:Rebekah Baskin, Meghanath Gali, Sung O. Park, Zhizhuang Joe Zhao, György M. Keserű, Kirpal S. Bisht, Peter P. Sayeski
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 3) pp:1402-1407
Publication Date(Web):1 February 2012
DOI:10.1016/j.bmcl.2011.12.042
In this study, we analyzed the structure–activity relationship properties of the small molecule Jak2 inhibitor G6. We synthesized a set of derivatives containing the native para-hydroxyl structure or an alternative meta-hydroxyl structure and examined their Jak2 inhibitory properties. We found that the para-hydroxyl derivative known as NB15 had excellent Jak2 inhibitory properties in silico, in vitro, and ex vivo when compared with meta-hydroxyl derivatives. These results indicate that NB15 is a potent derivative of the Jak2 inhibitor G6, and that maintaining the para-hydroxyl orientation of G6 is critical for its Jak2 inhibitory potential.
Co-reporter:Kavitha Gnanasambandan;Andrew T. Magis
Molecular and Cellular Biochemistry 2012 Volume 367( Issue 1-2) pp:125-140
Publication Date(Web):2012 August
DOI:10.1007/s11010-012-1326-7
Jak2 mutations in the exon 14 and exon 12 regions that cause constitutive activation have been associated with myeloproliferative neoplasms. We have previously shown that a pi stacking interaction between F617 and F595 is important for the constitutive activation of Jak2-V617F (Gnanasambandan et al., Biochemistry 49:9972–9984, 2010). Here, using a combination of molecular dynamics (MD) simulations and in vitro mutagenesis, we studied the molecular mechanism for the constitutive activation of the Jak2 exon 12 mutation, H538Q/K539L. The activation levels of Jak2-H538Q/K539L were found to be similar to that of Jak2-V617F, and Jak2-H538Q/K539L/V617F. Data from MD simulations indicated a shift in the salt bridge interactions of D620 and E621 with K539 in Jak2-WT to R541 in Jak2-H538Q/K539L. When compared to Jak2-WT, K539A mutation resulted in increased activation, while K539D or K539E mutations diminished Jak2 activation by 50 %. In the context of Jak2-H538Q/K539L, R541A mutation reduced its activation by 50 %, while R541D and R541E mutations returned its activation levels to that of Jak2-WT. Collectively, these results indicate that a shift in the salt bridge interaction of D620 and E621 with K539 in Jak2-WT to R541 in Jak2-H538Q/K539L is critical for constitutive activation of this Jak2 exon 12 mutant.
Co-reporter:Anurima Majumder, Annet Kirabo, Kanchana Karrupiah, Shigeharu Tsuda, Jennifer Caldwell-Busby, Arturo J. Cardounel, György M. Keserű, and Peter P. Sayeski
Biochemistry 2011 Volume 50(Issue 36) pp:
Publication Date(Web):August 8, 2011
DOI:10.1021/bi200847n
Hyperkinetic Jak2 tyrosine kinase signaling has been implicated in several human diseases including leukemia, lymphoma, myeloma, and the myeloproliferative neoplasms. Using structure-based virtual screening, we previously identified a novel Jak2 inhibitor named G6. We showed that G6 specifically inhibits Jak2 kinase activity and suppresses Jak2-mediated cellular proliferation. To elucidate the molecular and biochemical mechanisms by which G6 inhibits Jak2-mediated cellular proliferation, we treated Jak2-V617F expressing human erythroleukemia (HEL) cells for 12 h with either vehicle control or 25 μM of the drug and compared protein expression profiles using two-dimensional gel electrophoresis. One differentially expressed protein identified by electrospray mass spectroscopy was the intermediate filament protein, vimentin. It was present in DMSO treated cells but absent in G6 treated cells. HEL cells treated with G6 showed both time- and dose-dependent cleavage of vimentin as well as a marked reorganization of vimentin intermediate filaments within intact cells. In a mouse model of Jak2-V617F mediated human erythroleukemia, G6 also decreased the levels of vimentin protein, in vivo. The G6-induced cleavage of vimentin was found to be Jak2-dependent and calpain-mediated. Furthermore, we found that intracellular calcium mobilization is essential and sufficient for the cleavage of vimentin. Finally, we show that the cleavage of vimentin intermediate filaments, per se, is sufficient to reduce HEL cell viability. Collectively, these results suggest that G6-induced inhibition of Jak2-mediated pathogenic cell growth is concomitant with the disruption of intracellular vimentin filaments. As such, this work describes a novel pathway for the targeting of Jak2-mediated pathological cell growth.
Co-reporter:Kavitha Gnanasambandan, Andrew Magis, and Peter P. Sayeski
Biochemistry 2010 Volume 49(Issue 46) pp:
Publication Date(Web):October 19, 2010
DOI:10.1021/bi1014858
Somatic mutations in the Jak2 allele that lead to constitutive kinase activation of the protein have been identified in human disease conditions such as the myeloproliferative neoplasms (MPNs). The most common mutation in these patients is a V617F substitution mutation, which is believed to play a causative role in the MPN pathogenesis. As such, identifying the molecular basis for the constitutive activation of Jak2-V617F is important for understanding its clinical implications and potential treatment. Here, we hypothesized that conversion of residue 617 from Val to Phe resulted in the formation of novel π stacking interactions with neighboring Phe residues. To test this, we first examined the Jak2 structure via molecular modeling and identified a potential π stacking interaction between F594, F595, and F617. Disruption of this interaction through site-directed mutagenesis impaired Jak2 autophosphorylation, Jak2-dependent gene transcription, and in vitro kinase activity of the Jak2-V617F protein. Further, substitution of F594 and F595 with Trp did not affect Jak2 function significantly, but replacement with charged residues did, showing the importance of aromaticity and hydropathy index conservation at these positions. Using molecular dynamics (MD) simulations, we found that the π stacking interaction between residues 595 and 617 in the Jak2-V617F protein was of much greater energy and conformed to the properties of π stacking, relative to the Jak2-WT or Jak2-V617F/F594A/F595A. In summary, we have identified a π stacking interaction between F595 and F617 that is specific to and is critical for the constitutive activation of Jak2-V617F.
Co-reporter:Jacqueline Sayyah;Peter P. Sayeski
Current Oncology Reports 2009 Volume 11( Issue 2) pp:117-124
Publication Date(Web):2009 March
DOI:10.1007/s11912-009-0018-2
Although the Jak2-V617F mutation has generated strong awareness because of its causative role in myeloproliferative disorders, reports of Jak2 gene aberrations linked to hematologic malignancies have preceded those of V617F by nearly a decade. These malignant mutations include Jak2 amino acid substitutions, deletions, insertions, and chromosomal translocations. As a consequence, researchers are increasingly focused on identifying Jak2 inhibitors that suppress aberrant Jak2 kinase activity. Some of these inhibitors may one day become therapeutically beneficial for individuals with Jak2-related hematologic malignancies. This review summarizes various Jak2 mutations associated with hematologic malignancies and assesses some of the Jak2 inhibitors in the preclinical phase or in clinical trials. By reviewing these specific areas, we hope to have a better understanding of Jak2’s role in hematologic malignancies and to shed light on the utility of Jak2 inhibitors.
Co-reporter:Issam McDoom, Xianyue Ma, Annet Kirabo, Kuang-Yung Lee, David A. Ostrov and Peter P. Sayeski
Biochemistry 2008 Volume 47(Issue 32) pp:
Publication Date(Web):July 18, 2008
DOI:10.1021/bi800867d
Jak2 is a 130 kDa tyrosine kinase that is important in a number of cellular signaling pathways. Its function is intrinsically regulated by the phosphorylation of a handful of its 49 tyrosines. Here, we report that tyrosine 972 (Y972) is a novel site of Jak2 phosphorylation and, hence, autoregulation. Specifically, we found that Y972 is phosphorylated and confirmed that this residue resides on the surface of the protein. Using expression plasmids that expressed either wild-type Jak2 or a full-length Jak2 cDNA containing a single Y972F substitution mutation, we investigated the consequences of losing Y972 phosphorylation on Jak2 function. We determined that the loss of Y972 phosphorylation significantly reduced the levels of both Jak2 total tyrosine phosphorylation and phosphorylation of Y1007/Y1008. Additionally, Y972 phosphorylation was shown to be important for maximal kinase function. Interestingly, in response to classical cytokine activation, the Jak2 Y972F mutant exhibited a moderately impaired level of activation when compared to the wild-type protein. However, when Jak2 was activated via a GPCR ligand, the ability of the Y972F mutant to be activated was completely lost, therefore suggesting a differential role of Y972 in Jak2 activation. Finally, we found that phosphorylation of Y972 enhances Jak2 kinase function via a mechanism that appears to stabilize the active conformation of the protein. Collectively, our results suggest that Y972 is a novel site of Jak2 phosphorylation and plays an important differential role in ligand-dependent Jak2 activation via a mechanism that involves stabilization of the Jak2 active conformation.
Co-reporter:Annet Kirabo, Sung O. Park, Heather L. Wamsley, Meghanath Gali, Rebekah Baskin, Mary K. Reinhard, Zhizhuang J. Zhao, Kirpal S. Bisht, György M. Keserű, Christopher R. Cogle, Peter P. Sayeski
The American Journal of Pathology (September 2012) Volume 181(Issue 3) pp:
Publication Date(Web):1 September 2012
DOI:10.1016/j.ajpath.2012.05.033
Philadelphia chromosome–negative myeloproliferative neoplasms, including polycythemia vera, essential thrombocytosis, and myelofibrosis, are disorders characterized by abnormal hematopoiesis. Among these myeloproliferative neoplasms, myelofibrosis has the most unfavorable prognosis. Furthermore, currently available therapies for myelofibrosis have little to no efficacy in the bone marrow and hence, are palliative. We recently developed a Janus kinase 2 (Jak2) small molecule inhibitor called G6 and found that it exhibits marked efficacy in a xenograft model of Jak2-V617F–mediated hyperplasia and a transgenic mouse model of Jak2-V617F–mediated polycythemia vera/essential thrombocytosis. However, its efficacy in Jak2-mediated myelofibrosis has not previously been examined. Here, we hypothesized that G6 would be efficacious in Jak2-V617F–mediated myelofibrosis. To test this, mice expressing the human Jak2-V617F cDNA under the control of the vav promoter were administered G6 or vehicle control solution, and efficacy was determined by measuring parameters within the peripheral blood, liver, spleen, and bone marrow. We found that G6 significantly reduced extramedullary hematopoiesis in the liver and splenomegaly. In the bone marrow, G6 significantly reduced pathogenic Jak/STAT signaling by 53%, megakaryocytic hyperplasia by 70%, and the Jak2 mutant burden by 68%. Furthermore, G6 significantly improved the myeloid to erythroid ratio and significantly reversed the myelofibrosis. Collectively, these results indicate that G6 is efficacious in Jak2-V617F–mediated myelofibrosis, and given its bone marrow efficacy, it may alter the natural history of this disease.
Co-reporter:Annet Kirabo, Sung O. Park, Anurima Majumder, Meghanath Gali, ... Peter P. Sayeski
Neoplasia (November 2011) Volume 13(Issue 11) pp:1058-1068
Publication Date(Web):1 November 2011
DOI:10.1593/neo.111112
We recently developed a Janus kinase 2 (Jak2) small-molecule inhibitor called G6 and found that it inhibits Jak2-V617F– mediated pathologic cell growth in vitro, ex vivo, and in vivo. However, its ability to inhibit Jak2-V617F–mediated myeloproliferative neoplasia, with particular emphasis in the bone marrow, has not previously been examined. Here, we investigated the efficacy of G6 in a transgenic mouse model of Jak2-V617F–mediated myeloproliferative neoplasia. We found that G6 provided therapeutic benefit to the peripheral blood as determined by elimination of leukocytosis, thrombocytosis, and erythrocytosis. G6 normalized the pathologically high plasma concentrations of interleukin 6 (IL-6). In the liver, G6 eliminated Jak2-V617F–driven extramedullary hematopoiesis. With respect to the spleen, G6 significantly reduced both the spleno-megaly and megakaryocytic hyperplasia. In the critically important bone marrow, G6 normalized the pathologically high levels of phospho-Jak2 and phospho–signal transducer and activator of transcription 5 (STAT5). It significantly reduced the megakaryocytic hyperplasia in the marrow and completely normalized the M/E ratio. Most importantly, G6 selectively reduced the mutant Jak2 burden by 67% on average, with virtual elimination of mutant Jak2 cells in one third of all treated mice. Lastly, clonogenic assays using marrow stem cells from the myeloproliferative neoplasm mice revealed a time-dependent elimination of the clonogenic growth potential of these cells by G6. Collectively, these data indicate that G6 exhibits exceptional efficacy in the peripheral blood, liver, spleen, and, most importantly, in the bone marrow, thereby raising the possibility that this compound may alter the natural history of Jak2-V617F–mediated myeloproliferative neoplasia.
Co-reporter:Rebekah Baskin, Peter P. Sayeski
Cellular Signalling (February 2012) Volume 24(Issue 2) pp:435-442
Publication Date(Web):1 February 2012
DOI:10.1016/j.cellsig.2011.09.016
The serum- and glucocorticoid-inducible kinase 1 (SGK1) is known to regulate a wide variety of cellular processes, including renal sodium retention and cell survival. Angiotensin II (Ang II) is one of the many signaling molecules capable of regulating SGK1 expression, and is also known to impact cell survival. Here, we examined the role of SGK1 in Ang II-mediated cell survival. We hypothesized that Ang II protects cells from apoptosis by upregulating and activating SGK1. To test this, we examined the effects of Ang II stimulation on SGK1 expression and downstream signaling. We also examined the effects of Ang II treatment and siRNA-mediated SGK1 knockdown on apoptosis after serum starvation. We found that after 2 h of Ang II treatment, SGK1 mRNA expression was increased approximately 2-fold. This induction was sensitive to reductions in intracellular calcium levels after pretreatment with BAPTA-AM, but insensitive to the L-type calcium channel blocker verapamil. SGK1 induction was also sensitive to the tyrosine kinase inhibitor genistein. Ang II treatment also caused a rapid increase in the level of phosphorylation of SGK1 at Ser422 and Thr256, and Ser422 phosphorylation was rapamycin-sensitive. We found that Ang II treatment was protective against serum starvation-induced apoptosis, and this protective effect was significantly blunted when SGK1 was silenced via siRNA. Lastly, Ang II induced FOXO3A phosphorylation in an SGK1-dependent manner, thereby reducing the pro-apoptotic actions of FOXO3A. Overall, these results indicate that Ang II upregulates and activates SGK1, leading to increased cell survival via multiple, non-redundant mechanisms.Highlights► Ang II promotes cell survival through upregulation and activation of SGK1. ► Ang II increases the levels of SGK1 via calcium and tyrosine kinase signalling. ► Ang II increases the phosphorylation of SGK1 via an mTOR-dependent mechanism. ► SGK1 promotes cell survival by mediating the phosphorylation of FOXO3A at Thr32. ► These are novel mechanisms by which Ang II can mediate cell survival.
Co-reporter:Peter P. Sayeski
Regulatory Peptides (10 February 2014) Volume 189() pp:v-vi
Publication Date(Web):10 February 2014
DOI:10.1016/j.regpep.2014.03.002