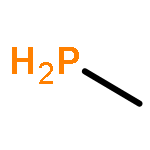
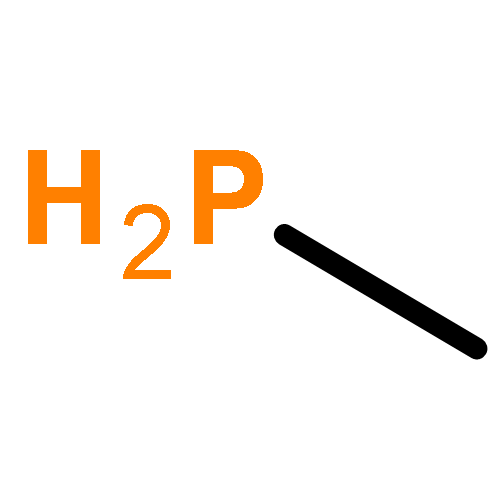
Co-reporter:Fang Liu;Likai Du;Zhenggang Lan
Photochemical & Photobiological Sciences (2002-Present) 2017 vol. 16(Issue 2) pp:211-219
Publication Date(Web):2017/02/15
DOI:10.1039/C6PP00367B
Sinapic acid derivatives are important sunscreen species in natural plants, which could provide protection from solar UV radiation. Using a combination of ultrafast excited state dynamics, together with classical molecular dynamics studies, we demonstrate that there is direct coupling of hydrogen bond motion with excited state photoprotection dynamics as part of the basic mechanism in solution. Beyond the intra-molecular degree of freedom, the inter-molecular motions on all timescales are potentially important for the photochemical or photophysical events, ranging from the ultrafast hydrogen bond motion to solvent rearrangements. This provides not only an enhanced understanding of the anomalous experimental spectroscopic results, but also the key idea in the development of sunscreen agents with improved photo-chemical properties. We suggest that the hydrogen bond dynamics coupled excited state photoprotection mechanism may also be possible in a broad range of bio-related molecules in the condensed phase.
Co-reporter:Qiang Wang, Jun Gao, Dongju Zhang, Chengbu Liu
Chemical Physics 2015 s 450–451() pp: 1-11
Publication Date(Web):1–15 April 2015
DOI:10.1016/j.chemphys.2015.01.011
•We theoretical studied peptide bond formation reaction mechanism with two water molecules.•The first water molecule can decrease the reaction barriers by forming hydrogen bonds.•The water molecule mediated three-proton transfer mechanism is the favorable mechanism.•Our calculation supports the two-step and eight membered ring mechanism.The ribosome is the macromolecular machine that catalyzes protein synthesis. The kinetic isotope effect analysis reported by Strobel group supports the two-step mechanism. However, the destination of the proton originating from the nucleophilic amine is uncertain. A computational simulation of different mechanisms including water molecules is carried out using the same reaction model and theoretical level. Formation the tetrahedral intermediate with proton transfer from nucleophilic nitrogen, is the rate-limiting step when two water molecules participate in peptide bond formation. The first water molecule forming hydrogen bonds with O9′ and H15′ in the A site can decrease the reaction barriers. Combined with results of the solvent isotope effects analysis, we conclude that the three-proton transfer mechanism in which water molecule mediate the proton shuttle between amino and carbon oxygen in rate-limiting step is the favorable mechanism. Our results will shield light on a better understand the reaction mechanism of ribosome.
Co-reporter:Lili Wang, Jun Gao, Fuzhen Bi, Bo Song, and Chengbu Liu
The Journal of Physical Chemistry A 2014 Volume 118(Issue 39) pp:9140-9147
Publication Date(Web):May 29, 2014
DOI:10.1021/jp502739c
As noncovalent intermolecular interactions, hydrogen bond (HB) and halogen bond (XB) are attracting increasing attention. In this work, the potential energy surfaces (PESs) of hydrogen and halogen bonds are compared. Twelve halogen-bonded and three hydrogen-bonded models are scanned for analysis using the MP2 level of theory. This work indicates that potential energy surfaces of both HB and XB have angular distortion. The potential well of XB is narrower than that of HB. With the elongation of the bond length, the potential energy surfaces get flatter. The best fitting functions for angular distortion and the flattening character of angular terms are also combined into a modified Buckingham potential. The testing results show that the essential features of the PES, including angular distortion and flattening character, have been reproduced. These results provide a better understanding of halogen and hydrogen bonds and the optimization of halogen bond force fields.
Co-reporter:Xin Che, Jun Gao, Yongjun Liu, Chengbu Liu
Journal of Inorganic Biochemistry 2013 Volume 122() pp:1-7
Publication Date(Web):May 2013
DOI:10.1016/j.jinorgbio.2013.01.008
Why the cysteine dioxygenase (CDO) cannot catalyze the oxidation of selenocysteine (Sec) but that of cysteine (Cys) is still an open question. In order to solve this question, the CDO model complex, the active site of CDO, and their selenium-substituted complexes have been selected as the computational models in this work. The stepwise donation of electron density during the first two reaction steps has been explored. In the first step, the electron density-donor ability of Se to donate to Fe is stronger than that of S; in the second step, S has the better electron density-donor ability to donate to O(2) than Se. Under the influence, in the Cys-bound complexes, the change of the oxidation state for the Fe center is II → III → II, while the Fe center in the Sec-bound complexes remains in the II oxidation state throughout. Considering that the ferric-superoxo species is an active oxidant and exhibits high reactivity in such reaction, it is speculated that the valence change of the Fe center makes the Cys-bound complexes effectively catalyze the oxidation of Cys, while the Sec-bound complexes cannot catalyze the oxidation of Sec. The competition for donation of electron density determines the valence change and the reaction ability.The competition of charge transfer determines the catalytic activity of cysteine dioxygenase and the valences of Fe center are different for selenocysteine and cysteine in reaction processes. This work explains why cysteine dioxygenase cannot catalyze the oxidation of selenocysteine but that of cysteine.Highlights► This work tries to understand the catalytic activity of cysteine dioxygenase for Sec and Cys. ► The competition of charge transfer determines the catalytic activity of cysteine dioxygenase. ► The valences of Fe center are different for Sec and Cys in reaction processes.
Co-reporter:Fuzhen Bi, Jun Gao, Lili Wang, Likai Du, Bo Song, Chengbu Liu
Chemical Physics 2013 Volume 426() pp:16-22
Publication Date(Web):29 November 2013
DOI:10.1016/j.chemphys.2013.09.006
Highlights
- •
A polarization-enhanced bonding process of halogen bonds is proposed.
- •
The contribution of polarization effects is 23.9% for the hydrogen bond dimer.
- •
The contribution of polarization effects varies from 2.5 to 50.5% for halogen bond dimers.
- •
The polarization and charge transfers are cooperated mutually.
Co-reporter:Likai Du, Jun Gao, Yongjun Liu, Dongju Zhang and Chengbu Liu
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 5) pp:1014-1024
Publication Date(Web):26 Oct 2011
DOI:10.1039/C1OB06221B
By employing ab initio quantum mechanical/molecular mechanical (QM/MM) and molecular dynamics (MD) simulations, we have provided further evidence against the previously proposed hydroperoxylation or hydroxylation mechanism of hydroxyethylphosphonate dioxygenase (HEPD). HEPD employs an interesting catalytic cycle based on concatenated bifurcations. The first bifurcation is based on the abstraction of hydrogen atoms from the substrate, which leads to a distal or proximal hydroperoxo species (Fe–OOH or Fe–(OH)O). The second and the third bifurcations refer to the carbon–carbon bond cleavage reaction. And this is achieved through a tridentate intermediate, or employing a proton-shuttle assisted mechanism, in which the residue Glu176 or the FeIVO group serves as a general base. The reaction directions seem to be tunable and show significant environment dependence. This mechanism can provide a comprehensive interpretation for the seemingly contradicting experimental evidences and provide insight into the development of biochemistry and material sciences.
Co-reporter:Qiang Wang, Jun Gao, Yongjun Liu, Chengbu Liu
Journal of Molecular Graphics and Modelling 2012 Volume 38() pp:186-193
Publication Date(Web):September 2012
DOI:10.1016/j.jmgm.2012.06.011
The protein tyrosine phosphatase 1B (PTP-1B) is acknowledged as an outstanding therapeutic target for the treatment of diabetes, obesity and cancer. In this work, six aryl diketoacid compounds have been studied on the basis of molecular dynamics simulations. Hydrogen bonds, binding energies and conformation changes of the WPD loop have been analyzed. The results indicated that their activation model falls into two parts: the target region of the monomeric aryl diketoacid compounds is the active site, whereas the target region of the dimeric aryl diketoacid compounds is the WPD loop or the R loop. The van der Waals interactions exhibit stronger effects than the short-range electrostatic interactions. The van der Waals interaction energy and the IC50 values exhibit an approximately exponential relationship. Furthermore, the van der Waals interactions cooperate with the hydrogen bond interactions. This study provides a more thorough understanding of the PTP-1B inhibitor binding processes.Graphical abstractHighlights► Monomeric and dimeric aryl diketoacids adopt different bonding modes. ► The van der Waals interaction plays an important role. ► The van der Waals interaction energy and their IC50 values are closely correlated. ► The van der Waals and the hydrogen bond interaction are mutually cooperative.
Co-reporter:Likai Du, Jun Gao, Yongjun Liu, and Chengbu Liu
The Journal of Physical Chemistry B 2012 Volume 116(Issue 39) pp:11837-11844
Publication Date(Web):September 5, 2012
DOI:10.1021/jp305454m
The hydroxyethylphosphonate dioxygenase (HEPD) catalyzes the critical carbon–carbon bond cleavage step in the phosphinothricin (PT) biosynthetic pathway. The experimental research suggests that water molecules play an important role in the catalytic reaction process of HEPD. This work proposes a water involved reaction mechanism where water molecules serve as an oxygen source in the generation of mononuclear nonheme iron oxo complexes. These molecules can take part in the catalytic cycle before the carbon–carbon bond cleavage process. The properties of trapped water molecules are also discussed. Meanwhile, water molecules seem to be responsible for converting the reactive hydroxyl radical group (−OH) to the ferric hydroxide (Fe(III)–OH) in a specific way. This converting reaction may prevent the enzyme from damages caused by the hydroxyl radical groups. So, water molecules may serve as biological catalysts just like the work in the heme enzyme P450 StaP. This work could provide a better interpretation on how the intermediates interact with water molecules and a further understanding on the O18 label experimental evidence in which only a relatively smaller ratio of oxygen atoms in water molecules (∼40%) are incorporated into the final product HMP.
Co-reporter:Xin Che, Jun Gao, Dongju Zhang, and Chengbu Liu
The Journal of Physical Chemistry A 2012 Volume 116(Issue 22) pp:5510-5517
Publication Date(Web):May 15, 2012
DOI:10.1021/jp3001515
In the iron(II)-thiolate models of cysteine dioxygenase, the thiolate ligand is a key factor in the oxygen activation. In this contribution, four model compounds have been theoretically investigated. This comparative study reveals that the thiolate ligand itself and its relative position are both important for the activation of O2. Before the O2 binding, the thiolate ligand must transfer charge to Fe(II), and the effective nuclear charges of Fe(II) is decreased, which results in a lower redox potential of compounds. In other words, the thiolate ligand provides a prerequisite for the O2 activation. Furthermore, the relative position of the thiolate ligand is discovered to determine the reaction path of O2 activation. The amount of charge transfer is crucial for these reactions; the more charge it transfers, the lower the related redox potentials. This work really helps think deeper into the O2 activation process of mononuclear nonheme iron enzymes.
Co-reporter:Qiang Wang, Jun Gao, Yongjun Liu, Chengbu Liu
Chemical Physics Letters 2010 Volume 501(1–3) pp:113-117
Publication Date(Web):6 December 2010
DOI:10.1016/j.cplett.2010.10.048
Abstract
A ribosome is a large RNA–protein machine that catalyzes protein synthesis. RNA plays a crucial role in catalyzing peptide bond formation. In this Letter, we focus on the catalytic role of the A76 2′-OH of the P site in tRNA. Three possible reaction schemes were compared using the same reaction model and theoretical level. By comparison of the potential energy surface and optimized geometries of transition states, a new favorable reaction pathway is shown. The activation energy is 24.0 kcal/mol. This work will provide a foundation for further theoretical and experimental study of ribosome mechanisms.
Co-reporter:Likai Du, Jun Gao, Yongjun Liu, Dongju Zhang and Chengbu Liu
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 5) pp:NaN1024-1024
Publication Date(Web):2011/10/26
DOI:10.1039/C1OB06221B
By employing ab initio quantum mechanical/molecular mechanical (QM/MM) and molecular dynamics (MD) simulations, we have provided further evidence against the previously proposed hydroperoxylation or hydroxylation mechanism of hydroxyethylphosphonate dioxygenase (HEPD). HEPD employs an interesting catalytic cycle based on concatenated bifurcations. The first bifurcation is based on the abstraction of hydrogen atoms from the substrate, which leads to a distal or proximal hydroperoxo species (Fe–OOH or Fe–(OH)O). The second and the third bifurcations refer to the carbon–carbon bond cleavage reaction. And this is achieved through a tridentate intermediate, or employing a proton-shuttle assisted mechanism, in which the residue Glu176 or the FeIVO group serves as a general base. The reaction directions seem to be tunable and show significant environment dependence. This mechanism can provide a comprehensive interpretation for the seemingly contradicting experimental evidences and provide insight into the development of biochemistry and material sciences.