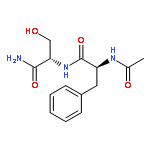
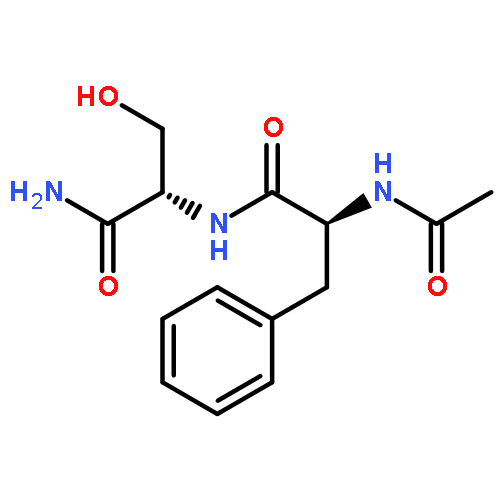
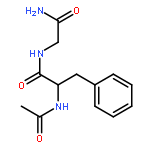
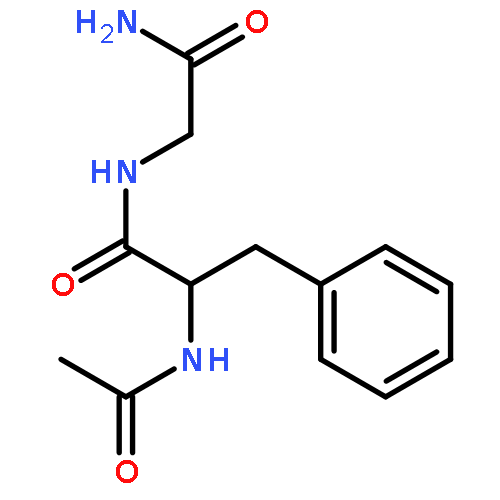
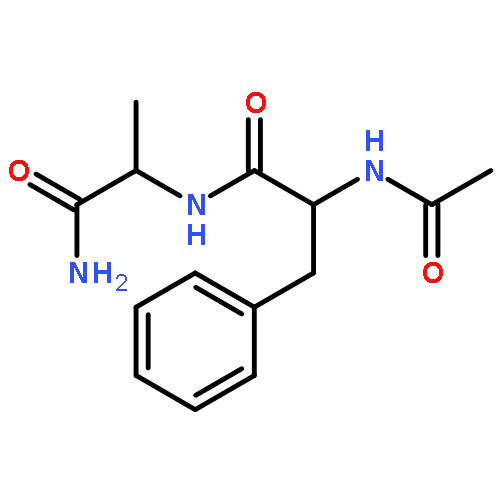
Co-reporter:Jérôme Mahé;Daniël J. Bakker;Sander Jaeqx;Marie-Pierre Gaigeot
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 21) pp:13778-13787
Publication Date(Web):2017/05/31
DOI:10.1039/C7CP00369B
Vibrational signatures of Ac-Phe-AA-NH2 dipeptides are recorded and analysed in the far IR/THz spectral domain (100–800 cm−1, 3–24 THz), with the ‘AA’ amino acid chosen within the series ‘AA’ = Gly, Ala, Pro, Cys, Ser, Val. Phe stands for phenylalanine. IR-UV ion dip experiments are conducted on the free electron laser FELIX and combined with DFT-based molecular dynamics simulations for the calculation of the dynamical anharmonic vibrational spectra. The excellent agreements between the experimental and theoretical spectra of the Ac-Phe-AA-NH2 series allow us to make detailed and unambiguous mapping of the vibrational motions into three main domains: 700–800 cm−1 for C–H waggings, 400–700 cm−1 for N–H waggings, with a one-to-one signature per amide N–H backbone group, 0–400 cm−1 for delocalized and large amplitude collective motions over the dipeptide backbone, with backbone torsional motions arising <100 cm−1.
Co-reporter:Daniël J. Bakker, Qin Ong, Arghya Dey, Jérôme Mahé, Marie-Pierre Gaigeot, Anouk M. Rijs
Journal of Molecular Spectroscopy 2017 Volume 342(Volume 342) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.jms.2017.02.004
•Gas phase far-infrared spectra of phenol and phenol derivatives.•Comparison of Born-Oppenheimer Molecular Dynamics and Static DFT.•Calculations of hydrogen bonding, flexibility, couplings and anharmonicity.•Coupling of functional groups and their influence on theories match to experiment.The far-infrared (far-IR) spectra of phenol and four ortho-substituted phenol derivatives, including three deuterated analogs, are presented. These spectra, measured using the free electron laser FELIX, are used to compare the performance of Born-Oppenheimer Molecular Dynamics (BOMD) with several commonly used levels of static density functional theory in the far-IR region. The molecules studied here form intramolecular hydrogen bonds of different strengths (except phenol), display diverse degrees of flexibility, and the OH moieties of the molecules provide large amplitude, anharmonic OH torsional modes. Since several of the molecules contain two OH groups, strong anharmonic couplings can also be present. Moreover, the experimental far-IR spectra of phenol and saligenin show overtones and combination bands as proven by the measurements of their deuterated analogs. All these characteristics of the molecules enable us to test the performance of the applied levels of theory on different complicating factors.Briefly summarized, both the strength of the hydrogen bond and molecular rigidity do not significantly influence the agreement between theory and experiment. All applied theoretical methods have difficulties to consistently predict modes that include the anharmonic OH torsional motion, resulting in overestimated intensities and frequencies. Coupling between two OH functional groups provides an additional challenge for theories, as seen for catechol. The various employed theoretical methods are found to complement each other, showing good results for complex harmonic modes in the case of static B3LYP-D3, while improved results are observed for anharmonic modes, including the OH torsional modes and their couplings, in the case of BOMD. Additionally, BOMD calculates the relative intensities better than the other theories. VPT2 reproduces weak anharmonic modes well, but it overestimates shifts and intensities for strong anharmonic modes.Download high-res image (133KB)Download full-size image
Co-reporter:Daniël J. Bakker;Arghya Dey;Daniel P. Tabor;Qin Ong;Jérôme Mahé;Marie-Pierre Gaigeot;Edwin L. Sibert, III
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 31) pp:20343-20356
Publication Date(Web):2017/08/09
DOI:10.1039/C7CP01951C
Saligenin (2-(hydroxymethyl)phenol) exhibits both strong and weak intramolecular electrostatic interactions. The bonds that result from these interactions compete with intermolecular hydrogen bonds once saligenin binds to one or more water molecules. Infrared (IR) ultraviolet (UV) ion-dip spectroscopy was used to study isolated saligenin–(H2O)n clusters (n = 1–3) in the far- and mid-IR regions of the spectrum. Both harmonic and anharmonic (coupled local modes and Born–Oppenheimer molecular dynamics) quantum chemical calculations were applied to assign cluster geometries to the measured spectra, and to assign vibrational modes to all spectral features measured for each cluster. The hydrated clusters with n = 1 and 2 have geometries that are quite similar to benzyl alcohol–water clusters, whereas the larger clusters with n = 3 show structures equivalent to the isolated water pentamer. Systematic shifts in the frequencies of three hydrogen bond (H-bond) deforming modes, namely OH stretching, OH torsion and H-bond stretching, were studied as a function of the hydrogen bond strength represented by either the OH bond length or the H-bond length. The shifts of the frequencies of these three modes correlate linearly to the OH length, despite both intra- and intermolecular H-bonds being included in this analysis. The OH torsion vibration displays the largest frequency shift when H-bonded, followed by the OH stretching vibrations and finally the H-bond stretching frequency. The frequency shifts of these H-bond deforming modes behave non-linearly as a function of the H-bond length, asymptotically approaching the frequency expected for the non H-bonded modes. The nonlinear behavior was quantified using exponential functions.
Co-reporter:P. Constantinidis, F. Hirsch, I. FischerA. Dey, A. M. Rijs
The Journal of Physical Chemistry A 2017 Volume 121(Issue 1) pp:
Publication Date(Web):December 8, 2016
DOI:10.1021/acs.jpca.6b08750
The propargyl radical is considered to be of key importance in the formation of the first aromatic ring in combustion processes. Here we study the bimolecular (self-) reactions of propargyl in a high-temperature pyrolysis flow reactor. The aromatic reaction products are identified by IR/UV ion dip spectroscopy, using the free electron laser FELIX as mid-infrared source. This technique combines mass selectivity with structural sensitivity. We identified several aromatic reaction products based on their infrared spectra, among them benzene, naphthalene, phenanthrene, indene, biphenyl, and surprisingly a number of aromatic compounds with acetylenic (ethynyl) side chains. The observation of benzene confirms that propargyl is involved in the formation of the first aromatic ring. The observation of compounds with acetylenic side chains shows that, in addition to a propargyl- and phenyl-based mechanism, the HACA (hydrogen abstraction C2H2 addition) mechanism of polycyclic aromatic hydrocarbons formation is present, although no acetylene was used as a reactant. On the basis of the experimental results we suggest a mechanism that connects the two pathways.
Co-reporter:Vasyl Yatsyna, Daniël J. Bakker, Raimund Feifel, Anouk M. Rijs and Vitali Zhaunerchyk
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 8) pp:6275-6283
Publication Date(Web):01 Feb 2016
DOI:10.1039/C5CP07426F
Spectroscopic studies of molecular structure can strongly benefit from extending the conventional mid-IR range to the far-IR and THz regions, as low-frequency molecular vibrations provide unique fingerprints and high sensitivity to intra- and intermolecular interactions. In this work, the gas-phase conformer specific far-IR spectra of aminophenol isomers, recorded in the spectral range of 220–800 cm−1 at the free-electron laser laboratory FELIX in Nijmegen (the Netherlands), are reported. Many distinct far-IR vibrational signatures which are specific for the molecular structure of the different aminophenol isomers are revealed and assigned. The observed far-IR transitions of the NH2 wagging (inversion) motion have been treated with a double-minimum harmonic well potential model that has enabled us to obtain the inversion barrier values. Moreover, we discuss the limitations and capability of conventional DFT frequency calculations to describe the far-IR vibrational modes.
Co-reporter:Daniël J. Bakker; Atze Peters; Vasyl Yatsyna; Vitali Zhaunerchyk
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 7) pp:1238-1243
Publication Date(Web):March 16, 2016
DOI:10.1021/acs.jpclett.6b00016
One of the most direct ways to study the intrinsic properties of the hydrogen-bond interaction is by gas-phase far-infrared (far-IR) spectroscopy because the modes involving hydrogen-bond deformation are excited in this spectral region; however, the far-IR regime is often ignored in molecular structure identification due to the absence of strong far-IR light sources and difficulty in assigning the observed modes by quantum chemical calculations. Far-IR/UV ion-dip spectroscopy using the free electron laser FELIX was applied to directly probe the intramolecular hydrogen-bond interaction in a family of phenol derivatives. Three vibrational modes have been identified, which are expected to be diagnostic for the hydrogen-bond strength: hydrogen-bond stretching and hydrogen-bond-donating and -accepting OH torsion vibrations. Their position is evaluated with respect to the hydrogen bond strength, that is, the length of the hydrogen-bonded OH length. This shows that the hydrogen bond stretching frequency is diagnostic for the size of the ring that is closed by the hydrogen bond, while the strength of the hydrogen bond can be determined from the hydrogen-bond-donating OH torsion frequency. The combination of these two normal modes allows the direct probing of intramolecular hydrogen-bond characteristics using conformation-selective far-IR vibrational spectroscopy.
Co-reporter:Jérôme Mahé, Sander Jaeqx, Anouk M. Rijs and Marie-Pierre Gaigeot
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 39) pp:25905-25914
Publication Date(Web):26 May 2015
DOI:10.1039/C5CP01518A
The combination of conformation selective far-IR/UV double resonance spectroscopy with Born–Oppenheimer molecular dynamics (BOMD) simulations is presented here for the structural characterization of the Ac-Phe-Pro-NH2 peptide in the far-infrared spectral domain, i.e. for radiation below 800 cm−1. Two conformers have been shown to be present in the experiment, namely a conformer with a γ-turn fold (C7 interaction) and a β-turn fold (C10 interaction). The combined experimental and theoretical work presented here aims to provide spectral features typical of each conformer in this far-IR domain. The simulated BOMD far-IR spectra agree well with the experimental spectra and allow direct assignment of the observed bands. These assignments show that the 400–550 cm−1 spectral domain is conformer selective, allowing us to distinguish the H-bond signature of the γ-turn from the β-turn.
Co-reporter:Bin Yan, Sander Jaeqx, Wim J. van der Zande and Anouk M. Rijs
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 22) pp:10770-10778
Publication Date(Web):23 Apr 2014
DOI:10.1039/C4CP00810C
The conformational preferences of peptides are mainly controlled by the stabilizing effect of intramolecular interactions. In peptides with polar side chains, not only the backbone but also the side chain interactions determine the resulting conformations. In this paper, the conformational preferences of the capped dipeptides Ac-Phe-Ser-NH2 (FS) and Ac-Phe-Cys-NH2 (FC) are resolved under laser-desorbed jet cooling conditions using IR-UV ion dip spectroscopy and density functional theory (DFT) quantum chemistry calculations. As serine (Ser) and cysteine (Cys) only differ in an OH (Ser) or SH (Cys) moiety; this subtle alteration allows us to study the effect of the difference in hydrogen bonding for an OH and SH group in detail, and its effect on the secondary structure. IR absorption spectra are recorded in the NH stretching region (3200–3600 cm−1). In combination with quantum chemical calculations the spectra provide a direct view of intramolecular interactions. Here, we show that both FS as FC share a singly γ-folded backbone conformation as the most stable conformer. The hydrogen bond strength of OH⋯O (FS) is stronger than that of SH⋯O (FC), resulting in a more compact gamma turn structure. A second conformer is found for FC, showing a β turn interaction.
Co-reporter:Ser Jaeqx;Dr. Jos Oomens;Dr. Alvaro Cimas;Dr. Marie-Pierre Gaigeot;Dr. Anouk M. Rijs
Angewandte Chemie International Edition 2014 Volume 53( Issue 14) pp:3663-3666
Publication Date(Web):
DOI:10.1002/anie.201311189
Abstract
Vibrational spectroscopy provides an important probe of the three-dimensional structures of peptides. With increasing size, these IR spectra become very complex and to extract structural information, comparison with theoretical spectra is essential. Harmonic DFT calculations have become a common workhorse for predicting vibrational frequencies of small neutral and ionized gaseous peptides.1 Although the far-IR region (<500 cm−1) may contain a wealth of structural information, as recognized in condensed phase studies,2 DFT often performs poorly in predicting the far-IR spectra of peptides. Here, Born–Oppenheimer molecular dynamics (BOMD) is applied to predict the far-IR signatures of two γ-turn peptides. Combining experiments and simulations, far-IR spectra can provide structural information on gas-phase peptides superior to that extracted from mid-IR and amide A features.
Co-reporter:Sander Jaeqx, Jos Oomens and Anouk M. Rijs
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 38) pp:16341-16352
Publication Date(Web):03 Sep 2013
DOI:10.1039/C3CP52508B
The gas-phase side chain–side chain (SC–SC) interaction and possible proton transfer between glutamic acid (Glu) and arginine (Arg) residues are studied under low-temperature conditions in an overall neutral peptide. Conformation-specific IR spectra, obtained with the free electron laser FELIX, in combination with density functional theory (DFT) calculations, provide insight into the occurrence of intramolecular proton transfer and detailed information on the conformational preferences of the peptides Z-Glu-Alan-Arg-NHMe (n = 0,1,3). Low-energy structures are obtained using molecular dynamics simulations via the simulated annealing approach, resulting in three types of SC–SC interactions, in particular two types of pair-wise interactions and one bifurcated interaction. These low-energy structures are optimized and frequency calculations are performed using the B3LYP functional, for structural analysis, and the M05-2x functional, for relative energies, employing the 6-311+G(d,p) basis set. Comparison of experimental and computed spectra suggests that only a single conformation was present for each of the three peptides. Despite the increasing spacing between the Glu and Arg residues, the peptides have several types of interactions in common, in particular specific SC–SC and dispersion interactions between the Arg side chain and the phenyl ring of the Z-cap. Comparison with previous experiments on Ac-Glu-Ala-Phe-Ala-Arg-NHMe as well as molecular dynamics simulations further suggest that the pairwise interaction observed here is indeed energetically most favorable for short peptide sequences.
Co-reporter:Sander Jaeqx, Weina Du, Evert Jan Meijer, Jos Oomens, and Anouk M. Rijs
The Journal of Physical Chemistry A 2013 Volume 117(Issue 6) pp:1216-1227
Publication Date(Web):October 24, 2012
DOI:10.1021/jp3053339
The gas-phase conformational preferences of the model dipeptides Z-Glu-OH and Z-Arg-OH have been studied in the low-temperature environment of a supersonic jet. IR-UV ion-dip spectra obtained using the free electron laser FELIX provide conformation-specific IR spectra, which in combination with density functional theory (DFT) allow us to determine the conformational structures of the peptides. Molecular dynamics modeling using simulated annealing generates a variety of low-energy structures, for which geometry optimization and frequency calculations are then performed using the B3LYP functional with the 6-311+G(d,p) basis set. By comparing experimental and theoretical IR spectra, three conformations for Z-Glu-OH and two for Z-Arg-OH have been identified. For three of the five structures, the dispersion interaction provides an important contribution to the stabilization, emphasizing the importance of these forces in small peptides. Therefore, dispersion-corrected DFT functionals (M05-2X and B97D) have also been employed in our theoretical analysis. Second-order Møller–Plesset perturbation theory (MP2) has been used as benchmark for the relative energies of the different conformational structures. Finally, we address the ongoing debate on the gas-phase structure of arginine by elucidating whether isolated arginine is canonical, tautomeric, or zwitterionic.
Co-reporter:Manuela Cirtog, Anouk M. Rijs, Yohan Loquais, Valérie Brenner, Benjamin Tardivel, Eric Gloaguen, and Michel Mons
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 22) pp:3307-3311
Publication Date(Web):October 26, 2012
DOI:10.1021/jz301440c
Far/mid-IR signatures of the first hydration step of a flexible biomolecule, the model peptide chain Ac-Phe-NH2, have been investigated in the gas phase using the selective IR/UV double-resonance laser technique. The broad spectral region investigated with the free-electron laser FELIX (150–800 cm–1/70–12 μm) provided a direct access to three intermolecular vibrational modes of monohydrates, in which the water molecule bridges neighboring NH and CO sites of the peptide backbone. The spectral features, analyzed with the help of quantum chemistry, are assigned to the IR activity of the libration and wagging motions of the water molecule together with a strongly mode- and conformer-dependent vibrational coupling between solute and solvent molecules. These resolved spectra obtained in a so far poorly documented spectral region provide benchmark data, which should enable theoreticians of molecular interactions to assess their methods, in terms of both intermolecular potentials and treatment of the vibrational anharmonicity.Keywords: far IR; hydration; inter- versus intramolecular coupling; intermolecular vibrations; mid-IR spectroscopy; peptide; spectroscopy;
Co-reporter:Kathrin H. Fischer, Jörg Herterich, Ingo Fischer, Sander Jaeqx, and Anouk M. Rijs
The Journal of Physical Chemistry A 2012 Volume 116(Issue 33) pp:8515-8522
Publication Date(Web):July 25, 2012
DOI:10.1021/jp306075a
Two C9H7 isomers, 1-phenylpropargyl and 3-phenylpropargyl, have been studied by IR/UV double resonance spectroscopy in a free jet. The species are possible intermediates in the formation of soot and polycyclic aromatic hydrocarbons (PAH). The radicals are generated by flash pyrolysis from the corresponding bromides and ionized at 255–297 nm in a one-color, two-photon process. Mid-infrared radiation between 500 and 1800 cm–1 is provided by a free electron laser (FEL). It is shown that the two radicals can be distinguished by their infrared spectra. In addition, we studied the dimerization products originating from the phenylpropargyl self-reaction. We utilize the fact that the pyrolysis tube can be considered to be a flow reactor permitting us to investigate the chemistry in such a thermal reactor. Dimerization of phenylpropargyl produces predominately species with m/z = 228 and 230. A surprisingly high selectivity has been found: The species with m/z = 230 is identified to be para-terphenyl, whereas m/z = 228 can be assigned to 1-phenylethynyl-naphthalene. The results allow to derive a mechanism for the dimerization of phenylpropargyl and suggest hitherto unexplored pathways to the formation of soot and PAH.
Co-reporter:Jérôme Mahé, Daniël J. Bakker, Sander Jaeqx, Anouk M. Rijs and Marie-Pierre Gaigeot
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 21) pp:NaN13787-13787
Publication Date(Web):2017/04/27
DOI:10.1039/C7CP00369B
Vibrational signatures of Ac-Phe-AA-NH2 dipeptides are recorded and analysed in the far IR/THz spectral domain (100–800 cm−1, 3–24 THz), with the ‘AA’ amino acid chosen within the series ‘AA’ = Gly, Ala, Pro, Cys, Ser, Val. Phe stands for phenylalanine. IR-UV ion dip experiments are conducted on the free electron laser FELIX and combined with DFT-based molecular dynamics simulations for the calculation of the dynamical anharmonic vibrational spectra. The excellent agreements between the experimental and theoretical spectra of the Ac-Phe-AA-NH2 series allow us to make detailed and unambiguous mapping of the vibrational motions into three main domains: 700–800 cm−1 for C–H waggings, 400–700 cm−1 for N–H waggings, with a one-to-one signature per amide N–H backbone group, 0–400 cm−1 for delocalized and large amplitude collective motions over the dipeptide backbone, with backbone torsional motions arising <100 cm−1.
Co-reporter:Bin Yan, Sander Jaeqx, Wim J. van der Zande and Anouk M. Rijs
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 22) pp:NaN10778-10778
Publication Date(Web):2014/04/23
DOI:10.1039/C4CP00810C
The conformational preferences of peptides are mainly controlled by the stabilizing effect of intramolecular interactions. In peptides with polar side chains, not only the backbone but also the side chain interactions determine the resulting conformations. In this paper, the conformational preferences of the capped dipeptides Ac-Phe-Ser-NH2 (FS) and Ac-Phe-Cys-NH2 (FC) are resolved under laser-desorbed jet cooling conditions using IR-UV ion dip spectroscopy and density functional theory (DFT) quantum chemistry calculations. As serine (Ser) and cysteine (Cys) only differ in an OH (Ser) or SH (Cys) moiety; this subtle alteration allows us to study the effect of the difference in hydrogen bonding for an OH and SH group in detail, and its effect on the secondary structure. IR absorption spectra are recorded in the NH stretching region (3200–3600 cm−1). In combination with quantum chemical calculations the spectra provide a direct view of intramolecular interactions. Here, we show that both FS as FC share a singly γ-folded backbone conformation as the most stable conformer. The hydrogen bond strength of OH⋯O (FS) is stronger than that of SH⋯O (FC), resulting in a more compact gamma turn structure. A second conformer is found for FC, showing a β turn interaction.
Co-reporter:Sander Jaeqx, Jos Oomens and Anouk M. Rijs
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 38) pp:NaN16352-16352
Publication Date(Web):2013/09/03
DOI:10.1039/C3CP52508B
The gas-phase side chain–side chain (SC–SC) interaction and possible proton transfer between glutamic acid (Glu) and arginine (Arg) residues are studied under low-temperature conditions in an overall neutral peptide. Conformation-specific IR spectra, obtained with the free electron laser FELIX, in combination with density functional theory (DFT) calculations, provide insight into the occurrence of intramolecular proton transfer and detailed information on the conformational preferences of the peptides Z-Glu-Alan-Arg-NHMe (n = 0,1,3). Low-energy structures are obtained using molecular dynamics simulations via the simulated annealing approach, resulting in three types of SC–SC interactions, in particular two types of pair-wise interactions and one bifurcated interaction. These low-energy structures are optimized and frequency calculations are performed using the B3LYP functional, for structural analysis, and the M05-2x functional, for relative energies, employing the 6-311+G(d,p) basis set. Comparison of experimental and computed spectra suggests that only a single conformation was present for each of the three peptides. Despite the increasing spacing between the Glu and Arg residues, the peptides have several types of interactions in common, in particular specific SC–SC and dispersion interactions between the Arg side chain and the phenyl ring of the Z-cap. Comparison with previous experiments on Ac-Glu-Ala-Phe-Ala-Arg-NHMe as well as molecular dynamics simulations further suggest that the pairwise interaction observed here is indeed energetically most favorable for short peptide sequences.
Co-reporter:Vasyl Yatsyna, Daniël J. Bakker, Raimund Feifel, Anouk M. Rijs and Vitali Zhaunerchyk
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 8) pp:NaN6283-6283
Publication Date(Web):2016/02/01
DOI:10.1039/C5CP07426F
Spectroscopic studies of molecular structure can strongly benefit from extending the conventional mid-IR range to the far-IR and THz regions, as low-frequency molecular vibrations provide unique fingerprints and high sensitivity to intra- and intermolecular interactions. In this work, the gas-phase conformer specific far-IR spectra of aminophenol isomers, recorded in the spectral range of 220–800 cm−1 at the free-electron laser laboratory FELIX in Nijmegen (the Netherlands), are reported. Many distinct far-IR vibrational signatures which are specific for the molecular structure of the different aminophenol isomers are revealed and assigned. The observed far-IR transitions of the NH2 wagging (inversion) motion have been treated with a double-minimum harmonic well potential model that has enabled us to obtain the inversion barrier values. Moreover, we discuss the limitations and capability of conventional DFT frequency calculations to describe the far-IR vibrational modes.
Co-reporter:Jérôme Mahé, Sander Jaeqx, Anouk M. Rijs and Marie-Pierre Gaigeot
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 39) pp:NaN25914-25914
Publication Date(Web):2015/05/26
DOI:10.1039/C5CP01518A
The combination of conformation selective far-IR/UV double resonance spectroscopy with Born–Oppenheimer molecular dynamics (BOMD) simulations is presented here for the structural characterization of the Ac-Phe-Pro-NH2 peptide in the far-infrared spectral domain, i.e. for radiation below 800 cm−1. Two conformers have been shown to be present in the experiment, namely a conformer with a γ-turn fold (C7 interaction) and a β-turn fold (C10 interaction). The combined experimental and theoretical work presented here aims to provide spectral features typical of each conformer in this far-IR domain. The simulated BOMD far-IR spectra agree well with the experimental spectra and allow direct assignment of the observed bands. These assignments show that the 400–550 cm−1 spectral domain is conformer selective, allowing us to distinguish the H-bond signature of the γ-turn from the β-turn.