Co-reporter:Duncan W. Bruce, Christopher P. Cabry, José N. Canongia Lopes, Matthew L. Costen, Lucía D’Andrea, Isabelle Grillo, Brooks C. Marshall, Kenneth G. McKendrick, Timothy K. Minton, Simon M. Purcell, Sarah Rogers, John M. Slattery, Karina Shimizu, Eric Smoll, and María A. Tesa-Serrate
The Journal of Physical Chemistry B June 22, 2017 Volume 121(Issue 24) pp:6002-6002
Publication Date(Web):May 1, 2017
DOI:10.1021/acs.jpcb.7b01654
Ionic-liquid (IL) mixtures hold great promise, as they allow liquids with a wide range of properties to be formed by mixing two common components rather than by synthesizing a large array of pure ILs with different chemical structures. In addition, these mixtures can exhibit a range of properties and structural organization that depend on their composition, which opens up new possibilities for the composition-dependent control of IL properties for particular applications. However, the fundamental properties, structure, and dynamics of IL mixtures are currently poorly understood, which limits their more widespread application. This article presents the first comprehensive investigation into the bulk and surface properties of IL mixtures formed from two commonly encountered ILs: 1-ethyl-3-methylimidazolium and 1-dodecyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([C2mim][Tf2N] and [C12mim][Tf2N]). Physical property measurements (viscosity, conductivity, and density) reveal that these IL mixtures are not well described by simple mixing laws, implying that their structure and dynamics are strongly composition dependent. Small-angle X-ray and neutron scattering measurements, alongside molecular dynamics (MD) simulations, show that at low mole fractions of [C12mim][Tf2N], the bulk of the IL is composed of small aggregates of [C12mim]+ ions in a [C2mim][Tf2N] matrix, which is driven by nanosegregation of the long alkyl chains and the polar parts of the IL. As the proportion of [C12mim][Tf2N] in the mixtures increases, the size and number of aggregates increases until the C12 alkyl chains percolate through the system and a bicontinuous network of polar and nonpolar domains is formed. Reactive atom scattering-laser-induced fluorescence experiments, also supported by MD simulations, have been used to probe the surface structure of these mixtures. It is found that the vacuum–IL interface is enriched significantly in C12 alkyl chains, even in mixtures low in the long-chain component. These data show, in contrast to previous suggestions, that the [C12mim]+ ion is surface active in this binary IL mixture. However, the surface does not become saturated in C12 chains as its proportion in the mixtures increases and remains unsaturated in pure [C12mim][Tf2N].
Co-reporter:Lewis M. Hall;Dr. David P. Tew;Dr. Natalie E. Pridmore;Dr. Adrian C. Whitwood;Dr. Jason M. Lynam;Dr. John M. Slattery
Angewandte Chemie International Edition 2017 Volume 56(Issue 26) pp:7551-7556
Publication Date(Web):2017/06/19
DOI:10.1002/anie.201702401
AbstractThe facile synthesis of a stable and isolable compound with a fluoroalkynyl group, M−C≡CF, is reported. Reaction of [Ru(C≡CH)(η5-C5Me5)(dppe)] with an electrophilic fluorinating agent (NFSI) results in the formation of the fluorovinylidene complex [Ru(=C=CHF)(η5-C5Me5)(dppe)][N(SO2Ph)2]. Subsequent deprotonation with LiN(SiMe3)2 affords the fluoroalkynyl complex [Ru(C≡CF)(η5-C5Me5)(dppe)]. In marked contrast to the rare and highly reactive examples of fluoroalkynes that have been reported previously, this compound can be readily isolated and structurally characterized. This has allowed the structure and bonding in the CCF motif to be explored. Further electrophilic fluorination of this species yields the difluorovinylidene complex [Ru(C=CF2)(η5-C5Me5)(dppe)][N(SO2Ph)2].
Co-reporter:Lucy M. Milner, Lewis M. Hall, Natalie E. Pridmore, Matthew K. Skeats, Adrian C. Whitwood, Jason M. Lynam and John M. Slattery
Dalton Transactions 2016 vol. 45(Issue 4) pp:1717-1726
Publication Date(Web):24 Dec 2015
DOI:10.1039/C5DT04596G
Metal vinylidene complexes are widely encountered, or postulated, as intermediates in a range of important metal-mediated transformations of alkynes. However, fluorovinylidene complexes have rarely been described and their reactivity is largely unexplored. By making use of the novel outer-sphere electrophilic fluorination (OSEF) strategy we have developed a rapid, robust and convenient method for the preparation of fluorovinylidene and trifluoromethylvinylidene ruthenium complexes from non-fluorinated alkynes. Spectroscopic investigations (NMR and UV/Vis), coupled with TD-DFT studies, show that fluorine incorporation results in significant changes to the electronic structure of the vinylidene ligand. The reactivity of fluorovinylidene complexes shows many similarities to non-fluorinated analogues, but also some interesting differences, including a propensity to undergo unexpected C–F bond cleavage reactions. Heating fluorovinylidene complex [Ru(η5-C5H5)(PPh3)2(CC{F}R)][BF4] led to C–H activation of a PPh3 ligand to form an orthometallated fluorovinylphosphonium ligand. Reaction with pyridine led to nucleophilic attack at the metal-bound carbon atom of the vinylidene to form a vinyl pyridinium species, which undergoes both C–H and C–F activation to give a novel pyridylidene complex. Addition of water, in the presence of chloride, leads to anti-Markovnikov hydration of a fluorovinylidene complex to form an α-fluoroaldehyde, which slowly rearranges to its acyl fluoride isomer. Therefore, fluorovinylidenes ligands may be viewed as synthetic equivalents of 1-fluoroalkynes providing access to reactivity not possible by other routes.
Co-reporter:Lucy M. Milner; Natalie E. Pridmore; Adrian C. Whitwood; Jason M. Lynam
Journal of the American Chemical Society 2015 Volume 137(Issue 33) pp:10753-10759
Publication Date(Web):August 13, 2015
DOI:10.1021/jacs.5b06547
Organofluorine chemistry plays a key role in materials science, pharmaceuticals, agrochemicals, and medical imaging. However, the formation of new carbon–fluorine bonds with controlled regiochemistry and functional group tolerance is synthetically challenging. The use of metal complexes to promote fluorination reactions is of great current interest, but even state-of-the-art approaches are limited in their substrate scope, often require activated substrates, or do not allow access to desirable functionality, such as alkenyl C(sp2)–F or chiral C(sp3)–F centers. Here, we report the formation of new alkenyl and alkyl C–F bonds in the coordination sphere of ruthenium via an unprecedented outer-sphere electrophilic fluorination mechanism. The organometallic species involved are derived from nonactivated substrates (pyridine and terminal alkynes), and C–F bond formation occurs with full regio- and diastereoselectivity. The fluorinated ligands that are formed are retained at the metal, which allows subsequent metal-mediated reactivity.
Co-reporter:Jason M. Lynam, Lucy M. Milner, Neetisha S. Mistry, John M. Slattery, Sally R. Warrington and Adrian C. Whitwood
Dalton Transactions 2014 vol. 43(Issue 11) pp:4565-4572
Publication Date(Web):17 Jan 2014
DOI:10.1039/C3DT52984C
The ruthenium naphthalene complex [Ru(η5-C5H5)(η6-C10H8)]+ is a catalyst precursor for the direct C–H alkenylation of pyridine and related nitrogen heterocycles by terminal alkynes. Stoichiometric studies have demonstrated that the naphthalene ligand may be displaced by either pyridine, 4-methylpyridine or dimethylaminopyridine (DMAP) to give species [Ru(η5-C5H5)L3]+ (L = nitrogen-based ligand). Reaction of in situ-generated [Ru(η5-C5H5)(py)3]+ (py = pyridine) with PPh3 results in the formation of [Ru(η5-C5H5)(PPh3)(py)2]+, the active catalyst for direct alkenylation, some [Ru(η5-C5H5)(PPh3)2(py)]+ is also formed in this reaction. A one-pot procedure is reported which has allowed for the nature of the nitrogen heterocycle and phosphine ligand to be evaluated. The sterically demanding phosphine PCy3 inhibits catalysis, and only trace amounts of product are formed when precursors containing a pentamethylcyclopentadienyl group were used. The greatest conversion was observed with PMe3 when used as co-ligand with [Ru(η5-C5H5)(η6-C10H8)]+.
Co-reporter:David G. Johnson ; Jason M. Lynam ; Neetisha S. Mistry ; John M. Slattery ; Robert J. Thatcher ;Adrian C. Whitwood
Journal of the American Chemical Society 2012 Volume 135(Issue 6) pp:2222-2234
Publication Date(Web):December 17, 2012
DOI:10.1021/ja3097256
A combined experimental and theoretical study has demonstrated that [Ru(η5-C5H5)(py)2(PPh3)]+ is a key intermediate, and active catalyst for, the formation of 2-substituted E-styrylpyridines from pyridine and terminal alkynes HC≡CR (R = Ph, C6H4-4-CF3) in a 100% atom efficient manner under mild conditions. A catalyst deactivation pathway involving formation of the pyridylidene-containing complex [Ru(η5-C5H5)(κ3-C3-C5H4NCH═CHR)(PPh3)]+ and subsequently a 1-ruthanaindolizine complex has been identified. Mechanistic studies using 13C- and D-labeling and DFT calculations suggest that a vinylidene-containing intermediate [Ru(η5-C5H5)(py)(═C═CHR)(PPh3)]+ is formed, which can then proceed to the pyridylidene-containing deactivation product or the desired product depending on the reaction conditions. Nucleophilic attack by free pyridine at the α-carbon in this complex subsequently leads to formation of a C–H agostic complex that is the branching point for the productive and unproductive pathways. The formation of the desired products relies on C–H bond cleavage from this agostic complex in the presence of free pyridine to give the pyridyl complex [Ru(η5-C5H5)(C5H4N)(═C═CHR)(PPh3)]. Migration of the pyridyl ligand (or its pyridylidene tautomer) to the α-carbon of the vinylidene, followed by protonation, results in the formation of the 2-styrylpyridine. These studies demonstrate that pyridylidene ligands play an important role in both the productive and nonproductive pathways in this catalyst system.
Co-reporter:Laura C. Forfar, Timothy J. Clark, Michael Green, Stephen M. Mansell, Christopher A. Russell, Rajashekharayya A. Sanguramath and John M. Slattery
Chemical Communications 2012 vol. 48(Issue 14) pp:1970-1972
Publication Date(Web):11 Jan 2012
DOI:10.1039/C2CC15291F
Reaction of equimolar quantities of MX (M = Au, Cu, X = Cl; M = Ag, X = OTf) and GaCl3 in CH2Cl2 with P4 leads to phosphorus ligating a cationic coinage metal centre. For Cu and Ag, ion-contacted coordination polymers are formed; for Au, an ion-separated complex is observed that features the [Au(η2-P4)2]+ cation, which is the first homoleptic Au–P4 complex to be characterised in the condensed phase.
Co-reporter:John M. Slattery and Sharifa Hussein
Dalton Transactions 2012 vol. 41(Issue 6) pp:1808-1815
Publication Date(Web):13 Dec 2011
DOI:10.1039/C1DT11636C
The fluoride ion affinities (FIAs) of 33 phosphenium ions with a range of substituents were calculated using ab inito and DFT methods. The use of these FIA data as a measure of the Lewis acidities of phosphenium ions is described and the FIAs of the species studied here are compared to FIA data for more commonly encountered Lewis acids. Phosphenium ions are often stronger Lewis acids than neutral species, but in many cases are less Lewis acidic than highly electrophilic cations such as [Me3C]+ or [Me3Si]+. The impact of mesomeric, inductive and steric substituent effects on FIAs are discussed and related to the underlying electronic structures of different cation types. A comparison between the FIAs of known “free” phosphenium ions with those that are currently unknown and other highly electrophilic cations suggests that some diaryl- and dialkylphosphenium ions may yet be accessible under the right conditions.
Co-reporter:Robert J. Thatcher;David G. Johnson;Dr. John M. Slattery;Dr. Richard E. Douthwaite
Chemistry - A European Journal 2012 Volume 18( Issue 14) pp:4329-4336
Publication Date(Web):
DOI:10.1002/chem.201103319
Abstract
Deprotonation of the 1-isopropyl-3-(phenylamino)pyridin-1-ium iodide gives the corresponding neutral betaine, which is formalised as a pyridinium-amido ligand when coordinated to a metal. Spectroscopic, structural and theoretical methods have been used to investigate the metal–ligand bonding, ligand dynamics and electron distribution. Collectively, the data show that the ligand can be characterised as a pseudo-amide and is a strong donor akin to alkyl phosphines and N-heterocyclic carbenes. Furthermore, rotation about both N substituent CN bonds occurs, which is in contrast to the two alternative pyridinium positional isomers that exhibit neutral resonance structures. For comparison, compounds and complexes derived from norharman were prepared, which contain an additional CC bond supporting conjugation and the accessibility of a neutral resonance structure. Notwithstanding the formal neutral structure, norharman-derived ligands are comparably strong donors, and have the additional advantage of exhibiting stability to dioxygen and water.
Co-reporter:David G. Johnson, Jason M. Lynam, John M. Slattery and Christine E. Welby
Dalton Transactions 2010 vol. 39(Issue 43) pp:10432-10441
Publication Date(Web):12 Oct 2010
DOI:10.1039/C0DT00431F
The ruthenium bis-acetate complex Ru(κ2-OAc)2(PPh3)2 reacts with HCCPh to afford the vinylidene-containing species Ru(κ1-OAc)(κ2-OAc)(CCHPh)(PPh3)2. An experimental study has demonstrated that this reaction occurs under very mild conditions, with significant conversion being observed at 255 K. At lower temperatures, evidence for a transient metallo-enol ester species Ru(κ1-OAc)(OC{Me}O–CCHPh)(PPh3)2 was obtained. A comprehensive theoretical study to probe the nature of the alkyne/vinylidene tautomerisation has been undertaken using Density Functional Theory. Calculations based on a number of isomers of the model system Ru(κ1-OAc)(κ2-OAc)(CCHMe)(PH3)2 demonstrate that both the η2(CC) alkyne complex Ru(κ1-OAc)(κ2-OAc)(η2-HCCMe)(PH3)2 and the C–H agostic σ-complex Ru(κ1-OAc)(κ2-OAc)(η2{CH}-HCCMe)(PH3)2 are minima on the potential energy surface. The lowest energy pathway for the formation of the vinylidene complex involves the intramolecular deprotonation of the σ-complex by an acetate ligand followed by reprotonation of the subsequently formed alkynyl ligand. This process is thus termed a Ligand-Assisted Proton Shuttle (LAPS). Calculations performed on the full experimental system Ru(κ1-OAc)(κ2-OAc)(CCHPh)(PPh3)2 reinforce the notion that lowest energy pathway involves the deprotonation/reprotonation of the alkyne by an acetate ligand. Inclusion of the full ligand substituents in the calculations are necessary to reproduce the experimental observation of Ru(κ1-OAc)(κ2-OAc)(CCHPh)(PPh3)2 as the thermodynamic product.
Co-reporter:David G. Johnson, Jason M. Lynam, John M. Slattery and Christine E. Welby
Dalton Transactions 2010 - vol. 39(Issue 43) pp:NaN10441-10441
Publication Date(Web):2010/10/12
DOI:10.1039/C0DT00431F
The ruthenium bis-acetate complex Ru(κ2-OAc)2(PPh3)2 reacts with HCCPh to afford the vinylidene-containing species Ru(κ1-OAc)(κ2-OAc)(CCHPh)(PPh3)2. An experimental study has demonstrated that this reaction occurs under very mild conditions, with significant conversion being observed at 255 K. At lower temperatures, evidence for a transient metallo-enol ester species Ru(κ1-OAc)(OC{Me}O–CCHPh)(PPh3)2 was obtained. A comprehensive theoretical study to probe the nature of the alkyne/vinylidene tautomerisation has been undertaken using Density Functional Theory. Calculations based on a number of isomers of the model system Ru(κ1-OAc)(κ2-OAc)(CCHMe)(PH3)2 demonstrate that both the η2(CC) alkyne complex Ru(κ1-OAc)(κ2-OAc)(η2-HCCMe)(PH3)2 and the C–H agostic σ-complex Ru(κ1-OAc)(κ2-OAc)(η2{CH}-HCCMe)(PH3)2 are minima on the potential energy surface. The lowest energy pathway for the formation of the vinylidene complex involves the intramolecular deprotonation of the σ-complex by an acetate ligand followed by reprotonation of the subsequently formed alkynyl ligand. This process is thus termed a Ligand-Assisted Proton Shuttle (LAPS). Calculations performed on the full experimental system Ru(κ1-OAc)(κ2-OAc)(CCHPh)(PPh3)2 reinforce the notion that lowest energy pathway involves the deprotonation/reprotonation of the alkyne by an acetate ligand. Inclusion of the full ligand substituents in the calculations are necessary to reproduce the experimental observation of Ru(κ1-OAc)(κ2-OAc)(CCHPh)(PPh3)2 as the thermodynamic product.
Co-reporter:Laura C. Forfar, Timothy J. Clark, Michael Green, Stephen M. Mansell, Christopher A. Russell, Rajashekharayya A. Sanguramath and John M. Slattery
Chemical Communications 2012 - vol. 48(Issue 14) pp:NaN1972-1972
Publication Date(Web):2012/01/11
DOI:10.1039/C2CC15291F
Reaction of equimolar quantities of MX (M = Au, Cu, X = Cl; M = Ag, X = OTf) and GaCl3 in CH2Cl2 with P4 leads to phosphorus ligating a cationic coinage metal centre. For Cu and Ag, ion-contacted coordination polymers are formed; for Au, an ion-separated complex is observed that features the [Au(η2-P4)2]+ cation, which is the first homoleptic Au–P4 complex to be characterised in the condensed phase.
Co-reporter:Jason M. Lynam, Lucy M. Milner, Neetisha S. Mistry, John M. Slattery, Sally R. Warrington and Adrian C. Whitwood
Dalton Transactions 2014 - vol. 43(Issue 11) pp:NaN4572-4572
Publication Date(Web):2014/01/17
DOI:10.1039/C3DT52984C
The ruthenium naphthalene complex [Ru(η5-C5H5)(η6-C10H8)]+ is a catalyst precursor for the direct C–H alkenylation of pyridine and related nitrogen heterocycles by terminal alkynes. Stoichiometric studies have demonstrated that the naphthalene ligand may be displaced by either pyridine, 4-methylpyridine or dimethylaminopyridine (DMAP) to give species [Ru(η5-C5H5)L3]+ (L = nitrogen-based ligand). Reaction of in situ-generated [Ru(η5-C5H5)(py)3]+ (py = pyridine) with PPh3 results in the formation of [Ru(η5-C5H5)(PPh3)(py)2]+, the active catalyst for direct alkenylation, some [Ru(η5-C5H5)(PPh3)2(py)]+ is also formed in this reaction. A one-pot procedure is reported which has allowed for the nature of the nitrogen heterocycle and phosphine ligand to be evaluated. The sterically demanding phosphine PCy3 inhibits catalysis, and only trace amounts of product are formed when precursors containing a pentamethylcyclopentadienyl group were used. The greatest conversion was observed with PMe3 when used as co-ligand with [Ru(η5-C5H5)(η6-C10H8)]+.
Co-reporter:John M. Slattery and Sharifa Hussein
Dalton Transactions 2012 - vol. 41(Issue 6) pp:NaN1815-1815
Publication Date(Web):2011/12/13
DOI:10.1039/C1DT11636C
The fluoride ion affinities (FIAs) of 33 phosphenium ions with a range of substituents were calculated using ab inito and DFT methods. The use of these FIA data as a measure of the Lewis acidities of phosphenium ions is described and the FIAs of the species studied here are compared to FIA data for more commonly encountered Lewis acids. Phosphenium ions are often stronger Lewis acids than neutral species, but in many cases are less Lewis acidic than highly electrophilic cations such as [Me3C]+ or [Me3Si]+. The impact of mesomeric, inductive and steric substituent effects on FIAs are discussed and related to the underlying electronic structures of different cation types. A comparison between the FIAs of known “free” phosphenium ions with those that are currently unknown and other highly electrophilic cations suggests that some diaryl- and dialkylphosphenium ions may yet be accessible under the right conditions.
Co-reporter:Lucy M. Milner, Lewis M. Hall, Natalie E. Pridmore, Matthew K. Skeats, Adrian C. Whitwood, Jason M. Lynam and John M. Slattery
Dalton Transactions 2016 - vol. 45(Issue 4) pp:NaN1726-1726
Publication Date(Web):2015/12/24
DOI:10.1039/C5DT04596G
Metal vinylidene complexes are widely encountered, or postulated, as intermediates in a range of important metal-mediated transformations of alkynes. However, fluorovinylidene complexes have rarely been described and their reactivity is largely unexplored. By making use of the novel outer-sphere electrophilic fluorination (OSEF) strategy we have developed a rapid, robust and convenient method for the preparation of fluorovinylidene and trifluoromethylvinylidene ruthenium complexes from non-fluorinated alkynes. Spectroscopic investigations (NMR and UV/Vis), coupled with TD-DFT studies, show that fluorine incorporation results in significant changes to the electronic structure of the vinylidene ligand. The reactivity of fluorovinylidene complexes shows many similarities to non-fluorinated analogues, but also some interesting differences, including a propensity to undergo unexpected C–F bond cleavage reactions. Heating fluorovinylidene complex [Ru(η5-C5H5)(PPh3)2(CC{F}R)][BF4] led to C–H activation of a PPh3 ligand to form an orthometallated fluorovinylphosphonium ligand. Reaction with pyridine led to nucleophilic attack at the metal-bound carbon atom of the vinylidene to form a vinyl pyridinium species, which undergoes both C–H and C–F activation to give a novel pyridylidene complex. Addition of water, in the presence of chloride, leads to anti-Markovnikov hydration of a fluorovinylidene complex to form an α-fluoroaldehyde, which slowly rearranges to its acyl fluoride isomer. Therefore, fluorovinylidenes ligands may be viewed as synthetic equivalents of 1-fluoroalkynes providing access to reactivity not possible by other routes.

![1H-Imidazolium, 1-methyl-3-[[3,4,5-tris(octyloxy)phenyl]methyl]-, chloride](http://img.cochemist.com/ccimg/666900/666856-71-9.png)
![1H-Imidazolium, 1-methyl-3-[[3,4,5-tris(octyloxy)phenyl]methyl]-, chloride](http://img.cochemist.com/ccimg/666900/666856-71-9_b.png)


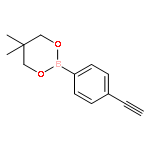
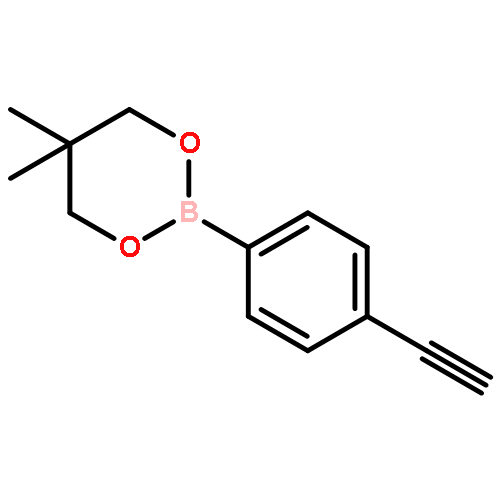
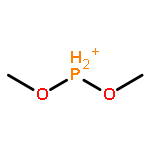

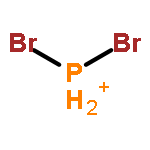

![2-[4-(5,5-dimethyl-1,3,2-dioxaborinan-2-yl)phenyl]ethynyl-trimethylsilane](http://img.cochemist.com/ccimg/183700/183677-72-7.png)
![2-[4-(5,5-dimethyl-1,3,2-dioxaborinan-2-yl)phenyl]ethynyl-trimethylsilane](http://img.cochemist.com/ccimg/183700/183677-72-7_b.png)





















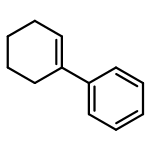
![2-[(e)-2-phenylethenyl]pyridine](http://img.cochemist.com/ccimg/600/538-49-8.png)
![2-[(e)-2-phenylethenyl]pyridine](http://img.cochemist.com/ccimg/600/538-49-8_b.png)


![Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, methyl ester, exo-](http://img.cochemist.com/ccimg/800/769-85-7.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, methyl ester, exo-](http://img.cochemist.com/ccimg/800/769-85-7_b.png)






![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/100/69-33-0.png)
![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/100/69-33-0_b.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, methyl ester, (1R,2R,4R)-rel-](/data/chemimg/1178900/2903-75-5.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, methyl ester, (1R,2R,4R)-rel-](/data/chemimg/1178900/2903-75-5_b.png)




![1-[4-(2-BROMOETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/55400/55368-24-6.png)
![1-[4-(2-BROMOETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/55400/55368-24-6_b.png)
















![2H-1-Benzopyran-2-one,4-hydroxy-3-[(1S)-3-oxo-1-phenylbutyl]-](http://img.cochemist.com/ccimg/5600/5543-57-7.png)
![2H-1-Benzopyran-2-one,4-hydroxy-3-[(1S)-3-oxo-1-phenylbutyl]-](http://img.cochemist.com/ccimg/5600/5543-57-7_b.png)


![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,5-iodo-7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/24400/24386-93-4.png)
![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,5-iodo-7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/24400/24386-93-4_b.png)