Co-reporter:Yuma Sakamoto, Takuaki Yamamoto, Yoshitomo Kajino, Tamon Kabata, ... Shiro Ikegawa
Journal of Orthopaedic Science 2017 Volume 22, Issue 5(Volume 22, Issue 5) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.jos.2016.01.010
Co-reporter:Aritoshi Iida, Nobuhiko Okamoto, Noriko Miyake, Gen Nishimura, Satoshi Minami, Takuya Sugimoto, Mitsuko Nakashima, Yoshinori Tsurusaki, Hirotomo Saitsu, Masaaki Shiina, Kazuhiro Ogata, Shigehiko Watanabe, Hirofumi Ohashi, Naomichi Matsumoto and Shiro Ikegawa
Journal of Human Genetics 2013 58(6) pp:391-394
Publication Date(Web):April 4, 2013
DOI:10.1038/jhg.2013.25
Opsismodysplasia is an autosomal recessive skeletal disorder characterized by facial dysmorphism, micromelia, platyspondyly and retarded bone maturation. Recently, mutations in the gene encoding inositol polyphosphate phosphatase-like 1 (INPPL1) are found in several families with opsismodysplasia by a homozygosity mapping, followed by whole genome sequencing. We performed an exome sequencing in two unrelated Japanese families with opsismodysplasia and identified a novel INPPL1 mutation, c.1960_1962delGAG, in one family. The mutation is predicted to result in an in-frame deletion (p.E654del) within the central catalytic 5-phosphate domain. Our results further support that INPPL1 is the disease gene for opsismodysplasia and that opsismodysplasia has genetic heterogeneity.
Co-reporter:Tatsuki Karasugi;Masahiro Nakajima
Journal of Bone and Mineral Metabolism 2013 Volume 31( Issue 2) pp:136-143
Publication Date(Web):2013 March
DOI:10.1007/s00774-012-0404-y
Ossification of the posterior longitudinal ligament of the spine (OPLL) is a common musculoskeletal disease among people after middle age. The OPLL presents with serious neurological abnormalities due to compression of the spinal cord and nerve roots. The OPLL is caused by genetic and environment factors; however, its etiology and pathogenesis still remain to be elucidated. To determine the susceptibility loci for OPLL, we performed a genome-wide linkage study using 214 affected sib-pairs of Japanese. In stratification analyses for definite cervical OPLL, we found loci with suggestive linkage on 1p21, 2p22–2p24, 7q22, 16q24 and 20p12. Fine mapping using additional markers detected the highest non-parametric linkage score (3.43, P = 0.00027) at D20S894 on chromosome 20p12 in a subgroup that had no complication of diabetes mellitus. Our result would shed a new light on genetic aspects of OPLL.
Co-reporter:Tae-Joon Cho;Kazu Matsumoto;Virginia Fano;Jin Dai;Ok-Hwa Kim;Jong Hee Chae;Won Joon Yoo;Yuji Tanaka;Yoshito Matsui;Iori Takigami;Soledad Monges;Bernhard Zabel;Katsuji Shimizu;Gen Nishimura;Ekkehart Lausch
American Journal of Medical Genetics Part A 2012 Volume 158A( Issue 4) pp:795-802
Publication Date(Web):
DOI:10.1002/ajmg.a.35268
Abstract
Heterozygous missense mutations of transient receptor potential vanilloid 4 channel (TRPV4) cause a spectrum of skeletal disorders, including brachyolmia, spondylometaphyseal dysplasia Kozlowski type, metatropic dysplasia, parastremmatic dysplasia, and spondyloepimetaphyseal dysplasia Maroteaux type. Similarly, heterozygous missense mutations of TRPV4 cause a spectrum of peripheral neuropathy, including hereditary motor and sensory neuropathy type IIC, congenital spinal muscular atrophy, and scapuloperoneal spinal muscular atrophy. There are no apparent differences in the amino acid positions affected or type of change predicted by the TRPV4 mutations responsible for the two disease spectrums; nevertheless, no fundamental phenotypic overlap has been shown between the two spectrums. Here, we report on three patients who had both skeletal dysplasia and peripheral neuropathy caused by heterozygous TRPV4 missense mutations. The skeletal and neurologic phenotypes of these patients covered the wide spectrum of reported TRPV4-pathies (disease caused by TRPV4 mutations). The molecular data are complementary, proving that “neuropathic” mutations can cause skeletal dysplasia but also the “skeletopathic” mutations can lead to neuropathies. Our findings suggest that pathogenic mechanisms of TRPV4-pathies in skeletal and nervous systems are not always mutually exclusive and provide further evidence that there is no clear genotype–phenotype correlation for either spectrum. Co-occurrence of skeletal dysplasia and degenerative neuropathy should be kept in mind in clinical practice including diagnostic testing, surgical evaluation, and genetic counseling. © 2012 Wiley Periodicals, Inc.
Co-reporter:Jin Dai, Ok-Hwa Kim, Tae-Joon Cho, Noriko Miyake, Hae-Ryong Song, Tatsuki Karasugi, Satoru Sakazume, Masahide Ikema, Yoshito Matsui, Toshiro Nagai, Naomichi Matsumoto, Hirofumi Ohashi, Naoyuki Kamatani, Gen Nishimura, Tatsuya Furuichi, Atsushi Takahashi and Shiro Ikegawa
Journal of Human Genetics 2011 56(5) pp:398-400
Publication Date(Web):March 17, 2011
DOI:10.1038/jhg.2011.28
Desbuquois dysplasia (DBQD) is a severe skeletal dysplasia of autosomal recessive inheritance. DBQD is classified into types 1 and 2 based on presence or absence of hand anomalies. In a previous study, we found a CANT1 (for calcium-activated nucleotidase 1) mutation, c.676G>A in five DBQD families. They were all East Asians (Japanese or Korean). The high prevalence of the same mutation among Japanese and Korean suggested that it is a common founder mutation in the two populations. To examine a possible common founder, we examined the region around CANT1 in chromosomes with c.676G>A mutation by genotyping polymorphic markers in the region for the families. We examined their haplotypes using the family data. We identified in all families a common haplotype containing the CANT1 mutation that ranged up to 550 kb. The two unrelated carriers of the mutation in general populations in Korea and Japan could also have the haplotype. We estimated the age of the founder mutation as ~1420 years (95% CI=880–1940 years). The c.676G>A mutation of CANT1 commonly seen in Japanese and Korean DBQD should be derived from a common founder.
Co-reporter:Masahiro Nakajima;Yoshinari Miyamoto
Journal of Bone and Mineral Metabolism 2011 Volume 29( Issue 3) pp:300-308
Publication Date(Web):2011 May
DOI:10.1007/s00774-010-0230-z
Osteoarthritis (OA) is one of the most prevalent skeletal diseases. Recently, we identified a novel gene on chromosome 3p24.3, named DVWA (double von Willebrand factor A domains), and its functional variants, which are associated with susceptibility to knee OA. Here we report the cloning and characterization of the DVWA gene. DVWA consisted of seven exons and had four alternative splicing variants, which encoded long (385 amino acid) and short (276 amino acid) proteins (L-DVWA and S-DVWA, respectively). S-DVWA was an N-terminal truncated form of L-DVWA and lacked a signal peptide and a part of a VWA domain. L-DVWA and S-DVWA transcripts were mainly expressed in articular cartilage. Immunoblot analysis using epitope-tagged proteins showed L-DVWA in the conditioned media and S-DVWA only in the cell, consistent with the in silico prediction. We also cloned the murine counterpart of DVWA, which was found to be identical to Col6a4, which has recently been reported. L-DVWA had 73% identity to the N-terminal sequence of the 2,309-amino acid Col6a4 protein. The mouse Dvwa/Col6a4 mRNA was present mainly in the small intestine in embryos and adults, but not in cartilage. The amino acid sequence of L-DVWA was conserved in higher species than chicken, but that of S-DVWA was unique in human. Knockdown of DVWA by siRNAs increased expression of chondrocyte matrix genes. Our study indicates that DVWA is evolutionally very unique, which, together with its specific expression in articular cartilage, suggests its specific role in human cartilage metabolism.
Co-reporter:Jin Dai and Shiro Ikegawa
Journal of Human Genetics 2010 55(2) pp:77-80
Publication Date(Web):January 15, 2010
DOI:10.1038/jhg.2009.137
Osteoarthritis (OA) is a polygenic disease with a definite genetic component, and recent advances in genome research have enabled us to investigate OA susceptibility genes. Several research groups, including ours, have reported the identification of OA susceptibility genes, mainly using candidate gene association studies. However, we are now entering the era of genome-wide association studies (GWAS). Here, we review recent progress in the study of susceptibility genes for OA, focusing in particular on GWAS and large-scale replication studies.
Co-reporter:Jin Dai, Tae-Joon Cho, Sheila Unger, Ekkehart Lausch, Gen Nishimura, Ok-Hwa Kim, Andrea Superti-Furga and Shiro Ikegawa
Journal of Human Genetics 2010 55(7) pp:400-402
Publication Date(Web):May 27, 2010
DOI:10.1038/jhg.2010.37
Transient receptor potential cation channel, subfamily V, member 4 (TRPV4) is a calcium-permeable nonselective cation channel of unknown biological function. TRPV4 mutation was first identified in brachyolmia, and then in a spectrum of autosomal-dominant skeletal dysplasias, which includes Kozlowski type of spondylometaphyseal dysplasia, metatropic dysplasia, Maroteaux type of spondyloepiphyseal dysplasia and parastremmatic dysplasia. Recently, TRPV4 mutation has also been identified in a spectrum of neuromuscular diseases that includes congenital distal spinal muscular atrophy (SMA), scapuloperoneal SMA, and hereditary motor and sensory neuropathy type IIC. These diverse spectrums of diseases compose a novel channelopathy, TRPV4-pathy, which could further include polygenic traits such as serum sodium concentration and a chronic obstructive pulmonary disease. In this review, we clarified the TRPV4 mutation spectrum, and discussed the phenotypic complexity of TRPV4-pathy and its pathogenic mechanisms. TRPV4-pathy may extend further to other monogenic and polygenic diseases.
Co-reporter:Ikuyo Kou;Masahiro Nakajima
Journal of Bone and Mineral Metabolism 2010 Volume 28( Issue 4) pp:395-402
Publication Date(Web):2010 July
DOI:10.1007/s00774-009-0145-8
Asporin is an extracellular matrix (ECM) protein that regulates cartilage matrix gene expression and cartilage formation by modulating the transforming growth factor-β (TGF-β) signaling pathway. Our previous studies have indicated that asporin binds to TGF-β1 directly and inhibits TGF-β1-mediated expression of cartilage matrix genes. However, it is still unknown how asporin interacts with TGF-β1 and influences its activity. Using competition assays, we determined that amino acids 159–205 of asporin mediate its interaction with TGF-β1 and effectively repress TGF-β1-induced cartilage matrix gene expression. Asporin also has a binding ability to type II collagen in vitro, but its binding pattern is different from that of TGF-β1. In contrast with previous in vivo findings, asporin did not affect the interaction between TGF-β1 and the TGF-β type II receptor (TβRII) by itself or in the presence of type II collagen in vitro. However, in the presence of heparin/heparan sulfate, asporin inhibits the interaction between TGF-β and TβRII in vitro. These findings suggest that asporin is one of the important cartilage matrix proteins that binds to the ECM and TGF-β1 and thereby modulates interactions between TGF-β and its signaling receptors.
Co-reporter:Qing Jiang;Dongquan Shi;Masahiro Nakajima;Jin Dai;Jia Wei
Journal of Human Genetics 2008 Volume 53( Issue 1) pp:
Publication Date(Web):2008 January
DOI:10.1007/s10038-007-0216-4
A genetic association of knee osteoarthritis (OA) and a C/T transition single nucleotide polymorphism (SNP) (rs912428) located in intron 1 of the LRCH1 gene has recently been reported in European Caucasians; however, the results are inconsistent. Our objective was to evaluate the association in different knee OA populations. Three case-control association studies were conducted in Han Chinese, Japanese, and Greek Caucasian populations. The LRCH1 SNP was genotyped in patients who had primary symptomatic knee OA with radiographic confirmation and in matched controls, and the association was examined. We performed a meta-analysis for the studies together with results of two previous papers using the DerSimonian–Laird procedure and calculated the power of the pooled studies by the software R. A total of 1,145 OA patients and 1,266 controls were genotyped. No significant difference was detected in genotype or allele frequencies between knee OA and control groups in the three populations (all P > 0.05). Association was not observed even after stratification by gender and Kellgren/Lawrence (K/L) scores. Meta-analysis also supported the lack of association between LRCH1 and knee OA. The strong heterogeneity between original and replication studies was detected in Caucasian populations. However, a tendency for the increase of TT genotype was observed in the European populations (OR = 1.46, P = 0.06). The powers for European and Asian replication studies were less than 0.8. Our results suggest that there is no association between LRCH1 and knee OA. However, lack of association should be concluded by further replication studies.
Co-reporter:Tatsuya Furuichi;Koichi Maeda;Chung-Tei Chou;Yu-Fen Liu
Journal of Human Genetics 2008 Volume 53( Issue 5) pp:419-424
Publication Date(Web):2008 May
DOI:10.1007/s10038-008-0265-3
Several genes have been implicated in the etiology of ankylosing spondylitis (AS); however, the significance of these genes except HLA-B27 remains to be elucidated. In this study, we examined the association of AS with novel candidate genes and previously reported genes other than HLA-B27. We examined a total of 45 single nucleotide polymorphisms (SNPs) in 15 genes by a sequential screening. We first genotyped 170 Japanese AS patients and 896 controls for the SNPs (first screen). Then, we genotyped eight SNPs with P < 0.05 in the first screen for 108 additional Japanese patients (second screen). We checked the replication of the association of the most significant SNP by genotyping 219 Taiwanese AS patients and 185 controls. When the first and second screens were combined, four SNPs showed nominal significance of P < 0.05. An intronic SNP (IVS1 + 996G > A) in MSX2, a novel candidate gene, showed the most significant association (P = 0.0030). The association was not replicated in our Taiwanese population; however, there was the same trend with the Japanese population in the allelic frequency distribution of the SNP. In the genes previously reported to have association with AS, only one synonymous SNP, c.963T > G in ANKH, showed a marginal association in the Japanese population (P = 0.045).
Co-reporter:Atsushi Miyake;Gen Nishimura;Toru Futami;Hirofumi Ohashi
Journal of Human Genetics 2008 Volume 53( Issue 8) pp:764-768
Publication Date(Web):2008 August
DOI:10.1007/s10038-008-0305-z
Diastrophic dysplasia sulfate transporter (DTDST) is required for synthesis of sulfated proteoglycans in cartilage, and its loss-of-function mutations result in recessively inherited chondrodysplasias. The 40 or so DTDST mutations reported to date cause a group of disorders termed the diastrophic dysplasia (DTD) group. The group ranges from the mildest recessive form of multiple epiphyseal dysplasia (r-MED) through the most common DTD to perinatally lethal atelosteogenesis type II and achondrogenesis 1B. Furthermore, the relationship between DTDST mutations, their sulfate transport function, and disease phenotypes has been described. Here we report a girl with DTDST mutations: a compound heterozygote of a novel p.T266I mutation and a recurrent p.ΔV340 mutation commonly found in severe phenotypes of the DTD group. In infancy, the girl presented with skeletal manifestations reminiscent of Desbuquois dysplasia, another recessively inherited chondrodysplasia, the mutations of which have never been identified. Her phenotype evolved with age into an intermediate phenotype between r-MED and DTD. Considering her clinical phenotypes and known phenotypes of p.ΔV340, p.T266I was predicted to be responsible for mild phenotypes of the DTD group. Our results further extend the phenotypic spectrum of DTDST mutations, adding Desbuquois dysplasia to the list of differential diagnosis of the DTD group.
Co-reporter:Dongquan Shi;Takahiro Nakamura;Masahiro Nakajima;Jin Dai
Arthritis Research & Therapy 2008 Volume 10( Issue 3) pp:
Publication Date(Web):2008 June
DOI:10.1186/ar2423
Conflicting findings on the association of single nucleotide polymorphisms (SNPs) in RHOB and TXNDC3 with susceptibility to knee osteoarthritis (OA) have been reported in European Caucasians. To examine the associations of these SNPs with OA in East Asian populations and to evaluate their global significance, we conducted two case-control studies in 955 Chinese and 750 Japanese patients.We genotyped the previously implicated SNPs rs585017 (in RHOB) and rs4720262 (in TXNDC3) in patients with primary symptomatic knee OA with radiographic confirmation and in matched control individuals, and analyzed their associations. We further conducted a meta-analysis of the study findings together with those of previously reported European studies using the DerSimonian-Laird procedure.A significant association of RHOB with knee OA was observed in male Chinese patients (P = 0.02). No significant associations were found for RHOB in any other comparisons in the East Asian populations. The association of TXNDC3 was replicated in Chinese female (P = 0.04) and Japanese (P = 0.03) patients, although none of these associations persisted after Bonferroni correction. Significant association (P = 0.02 for the allelic frequency) with nonsignificant heterogeneity was found in the East Asian replication study. No significant association was found in any comparison in the meta-analysis for all studies.Our study replicates the association, previously reported in European Caucasians, of TXNDC3 with knee OA susceptibility in an East Asian population.
Co-reporter:Masahiro Nakajima, Nobuhiko Haga, Kazuharu Takikawa, Noriyo Manabe, Gen Nishimura and Shiro Ikegawa
Journal of Human Genetics 2007 52(5) pp:473-475
Publication Date(Web):March 10, 2007
DOI:10.1007/s10038-007-0128-3
Fibrodysplasia ossificans progressiva (FOP) is a rare autosomal dominant disorder of skeletal malformations and presents progressive extra-skeletal ossification. The 617G>A (R206H) mutation in the activin receptor type IA (ACVR1) gene has been identified in all examined individuals with FOP of various ethnic groups, including Caucasian and Chinese descents. Here, we examined three Japanese patients with FOP for ACVR1 mutations. We identified the 617G>A mutation in all three patients. Our results suggest that the mutation in the ACVR1 gene is common and recurrent in the global population.
Co-reporter:Eiji Nakashima;Joseph R. Tran;Tim J.M. Welting;Ger J.M. Pruijn;Yuichiro Hirose;Gen Nishimura;Hirofumi Ohashi;Shepherd H. Schurman;Jun Cheng;Fabio Cotti;Ramaiah Nagaraja;David Schlessinger
American Journal of Medical Genetics Part A 2007 Volume 143A(Issue 22) pp:
Publication Date(Web):15 OCT 2007
DOI:10.1002/ajmg.a.32053
Cartilage hair hypoplasia (CHH; MIM 250250) is an autosomal recessive disease with diverse clinical manifestations. It is caused by mutations in RMRP gene, the RNA component of the ribonucleoprotein complex RNase MRP. Mutations in RMRP have been found in patients in the core promoter region or in the transcribed region, but the pathogenetic effect of the mutations is unclear. Real-time PCR assays confirmed that both promoter (c.−16_−1 dup and c.−15_+2 dup) and transcribed mutations (c.168G > A and c.218A > G) lower the expression level of RMRP. Experiments with 5′RACE, showed that the reduced transcription in the promoter mutants was accompanied by shifting of the transcription initiation sites to nucleotides 5′-upstream of the authentic site. Low levels of RMRP expression levels with transcript mutations were also seen when constructs encoding the wild-type and mutant genes were transfected into cultured cells. The reduced transcription was correlated with greater instability of mutant RMRP transcripts compared to controls. A comparable reduction was seen when a mouse gene containing the c.70A > G mutation (the major mutation in humans with CHH) was introduced into ES cells in place of one of the wild-type alleles. The low expression level of the c.70A > G Rmrp RNA was confirmed by expression assays into cultured cells, and was again correlated with RNA instability. Our results indicate that a loss of mutant RNA transcripts is a critical feature of pathogenesis. © 2007 Wiley-Liss, Inc.
Co-reporter:Yoshinari Miyamoto;Tatsuo Matsuda;Hiroshi Kitoh;Nobuhiko Haga
Human Genetics 2007 Volume 121( Issue 5) pp:625-629
Publication Date(Web):2007 June
DOI:10.1007/s00439-007-0354-y
Legg-Calvé-Perthes disease (LCPD) is a common childhood hip disorder characterized by sequential stages of involvement of the capital femoral epiphyses, including subchondral fracture, fragmentation, re-ossification and healing with residual deformity. Most cases are sporadic, but familial cases have been described, with some families having multiple affected members. Genetic factors have been implicated in the etiology of LCPD, but the causal gene has not been identified. We have located a missense mutation (p.G1170S) in the type II collagen gene (COL2A1) in a Japanese family with an autosomal dominant hip disorder manifesting as LCPD and showing considerable intra-familial phenotypic variation. This is the first report of a mutation in hereditary LCPD. COL2A1 mutations may be more common in LCPD patients than currently thought, particularly in familial and/or bilateral cases.
Co-reporter:Taichi Itoh;Shuya Shirahama;Eiji Nakashima;Koichi Maeda;Nobuhiko Haga;Hiroshi Kitoh;Rika Kosaki;Hirofumi Ohashi;Gen Nishimura
American Journal of Medical Genetics Part A 2006 Volume 140A(Issue 12) pp:
Publication Date(Web):11 MAY 2006
DOI:10.1002/ajmg.a.31292
Multiple epiphyseal dysplasia (MED) is among the most genetically heterogeneous skeletal dysplasias. Six genes involved in MED, COMP, MATN3, COL9A1, COL9A2, COL9A3, and DTDST have been identified; however, the presence of additional disease genes has been reported, and the detection rate for mutations in known genes accounts for no more than 50% of patients with MED in Western populations. Here, we screened the six known disease genes in 35 consecutive Japanese MED patients. We analyzed the entire coding region of each gene, along with flanking intron–exon junctions, by direct sequencing. A total of 19 mutations were identified in COMP, MATN3, COL9A2, COL9A3, and DTDST. The detection rate for known mutations was higher in this study than in previous reports, and we identified a substantially different spectrum of mutations. Mutations in MATN3 were more prevalent among these Japanese patients, whereas no DTDST mutations were detected. Most of the mutations were localized within specific regions of each gene: COMP mutations were found in the calmodulin-like repeat domains; MATN3 mutations in the von Willebrand factor type A domain; and type IX collagen gene mutations occurred in the third collagenous domains. Based on the integration of clinical and genetic information, we propose an algorithm for detecting mutations in Japanese MED patients. Our study further supports the existence of additional MED gene(s). © 2006 Wiley-Liss, Inc.
Co-reporter:Koichi Maeda;Yoshinari Miyamoto;Hideaki Sawai;Lawrence P. Karniski;Eiji Nakashima;Gen Nishimura
American Journal of Medical Genetics Part A 2006 Volume 140A(Issue 11) pp:
Publication Date(Web):26 APR 2006
DOI:10.1002/ajmg.a.31225
Diastrophic dysplasia sulfate transporter (DTDST) is a sulfate transporter required for the synthesis of sulfated proteoglycans in the cartilage. Over 30 mutations have been described in the DTDST gene, which result in a continuous clinical spectrum of recessively inherited chondrodysplasias, including, in order of increasing severity, a recessive form of multiple epiphyseal dysplasia (rMED), diastrophic dysplasia (DTD), atelosteogenesis type II (AO-II) and achondrogenesis 1B (ACG-1B). Correlation between disease severity and residual sulfate transport activity has been reported. Here we report a patient with DTDST mutations, whose manifestations fell in a range between AO-II and DTD. The patient was a compound heterozygote for the recurrent c.835C>T (p.R279W) and novel c.1987G>A (p.G663R) mutations. Immunocytochemical analysis in HEK293 cells showed that the p.G663R mutation was localized within the cytoplasm, and not to the cell membrane, suggesting p.G663R is a loss-of-function mutation. Our case supports the previously described correlation between the severity of the phenotype and the putative level of residual transport function. © 2006 Wiley-Liss, Inc.
Co-reporter:Yuichiro Hirose, Eiji Nakashima, Hirofumi Ohashi, Hiroshi Mochizuki, Yuki Bando, Tsutomu Ogata, Masanori Adachi, Emi Toba, Gen Nishimura and Shiro Ikegawa
Journal of Human Genetics 2006 51(8) pp:706-710
Publication Date(Web):July 11, 2006
DOI:10.1007/s10038-006-0015-3
Cartilage-hair hypoplasia (CHH), or metaphyseal dysplasia, McKusick type, is an autosomal recessive disease with diverse clinical manifestations. CHH is caused by mutations in RMRP (ribonuclease mitochondrial RNA processing), the gene encoding the RNA component of the ribonucleoprotein complex RNase MRP. A common founder mutation, 70A>G has been reported in the Finnish and Amish populations. We screened 11 Japanese patients with CHH for RMRP mutations and identified mutations in five probands, including three novel mutations (16-bp dup at +1, 168G>A, and 217C>T). All patients were compound heterozygotes for an insertion or duplication in the promoter or 5'-transcribed regions and a point mutation in the transcribed region. Two recurrent mutations were unique to the Japanese population: a 17-bp duplication at +3 and 218A>G. Haplotype analysis revealed that the two mutations common in Japanese individuals were contained within distinct haplotypes. Through this analysis, we have identified a unique mutation spectrum and founder mutations in the Japanese population.
Co-reporter:Shoji Seki, Yoshiharu Kawaguchi, Masaki Mori, Futoshi Mio, Kazuhiro Chiba, Yasuo Mikami, Tatsuhiko Tsunoda, Toshikazu Kubo, Yoshiaki Toyama, Tomoatsu Kimura and Shiro Ikegawa
Journal of Human Genetics 2006 51(12) pp:1063-1067
Publication Date(Web):September 23, 2006
DOI:10.1007/s10038-006-0062-9
Lumbar disc disease (LDD) is a common musculo-skeletal disease with strong genetic determinants. In a Finnish population, a single nucleotide polymorphism (SNP) causing an amino-acid substitution (Trp2 allele) in COL9A2, which encodes the 2 (IX) chain of type IX collagen, has been reported to associate with LDD. However, replication studies in different populations have produced controversial results. To further investigate the association of COL9A2 with LDD in Japanese, we examined SNPs in COL9A2, including Trp2, in 470 LDD patients (mean age 35) along with 658 controls (mean age 48). We identified a total of 43 sequence variations in COL9A2. Nine SNPs, including Trp2, were selected and genotyped. After Bonferroni's correction, none of these SNPs showed association. Unlike observations in the Finnish population, Trp2 was common in Japanese, and no association with LDD was apparent. However, we did see association of a COL9A2 specific haplotype with LDD (P=0.025; permutation test); this association is more significant in patients with severe lumbar disc degeneration (P=0.011). Thus, the association of Trp2 with LDD was not replicated, but COL9A2 susceptibility allele(s) other than Trp2 may be present in Japanese LDD.
Co-reporter:Shiro Ikegawa
Journal of Human Genetics 2006 51(7) pp:581-586
Publication Date(Web):May 3, 2006
DOI:10.1007/s10038-006-0401-x
Skeletal dysplasia is a group of disorders of the skeleton that result from derangement of growth, development and/or differentiation of the skeleton. Nearly 300 disorders are included; most of them are monogenic diseases. Responsible genes for skeletal dysplasia have been identified in more than 150 diseases mainly through positional cloning. Identification of disease genes would improve patient care through genetic diagnosis as well as improving our understanding of the diseases and molecular mechanism of skeletal tissue formation. Studies of skeletal dysplasia would also help identify disease genes for common diseases affecting bones and joints. In this study, the author reviews recent advances and the current status of the genetic analysis of skeletal dysplasia and its impacts on research into skeletal biology.
Co-reporter:
Nature Genetics 2005 37(6) pp:607-612
Publication Date(Web):01 May 2005
DOI:10.1038/ng1557
Lumbar disc disease (LDD) is caused by degeneration of intervertebral discs of the lumbar spine. One of the most common musculoskeletal disorders1, LDD has strong genetic determinants2,
3,
4. Using a case-control association study, we identified a functional SNP (1184T C, resulting in the amino acid substitution I395T) in CILP, which encodes the cartilage intermediate layer protein, that acts as a modulator of LDD susceptibility. CILP was expressed abundantly in intervertebral discs, and its expression increased as disc degeneration progressed. CILP colocalized with TGF-1 in clustering chondrocytes and their territorial matrices in intervertebral discs. CILP inhibited TGF-1−mediated induction of cartilage matrix genes through direct interaction with TGF-1 and inhibition of TGF-1 signaling. The susceptibility-associated 1184C allele showed increased binding and inhibition of TGF-1. Therefore, we conclude that the extracellular matrix protein CILP regulates TGF- signaling and that this regulation has a crucial role in the etiology and pathogenesis of LDD. Our study also adds to the list of connective tissue diseases that are associated with TGF-.
Co-reporter:
Nature Genetics 2005 37(2) pp:138 - 144
Publication Date(Web):09 January 2005
DOI:10.1038/ng1496
Co-reporter:Koichi Maeda;Eiji Nakashima;Taizo Horikoshi;Akihiko Mabuchi
American Journal of Medical Genetics Part A 2005 Volume 136A(Issue 3) pp:
Publication Date(Web):9 JUN 2005
DOI:10.1002/ajmg.a.30832
Co-reporter:Eiji Nakashima;Hirofumi Ohashi;Mamori Kimizuka;Gen Nishimura
American Journal of Medical Genetics Part A 2005 Volume 133A(Issue 1) pp:
Publication Date(Web):4 JAN 2005
DOI:10.1002/ajmg.a.30481
Co-reporter:Eiji Nakashima;Hiroshi Kitoh;Koichi Maeda;Nobuhiko Haga;Rika Kosaki;Akihiko Mabuchi;Gen Nishimura;Hirofumi Ohashi
American Journal of Medical Genetics Part A 2005 Volume 132A(Issue 2) pp:
Publication Date(Web):18 NOV 2004
DOI:10.1002/ajmg.a.30411
Multiple epiphyseal dysplasia (MED) is a common skeletal dysplasia characterized by mild to moderate short stature, early-onset of osteoarthritis (OA) mainly in the hip and knee joints, and abnormally small and/or irregular epiphyses. MED is clinically and genetically heterogeneous. Six causative genes of MED have been reported, including type IX collagen genes (COL9A1, COL9A2, COL9A3). All the type IX collagen mutations previously reported cause exon skipping that loses the COL3 domain. Here we have identified a novel COL9A3 mutation co-segregating in a three-generation family with MED. The mutation (IVS3 + 5G > A) was speculated to lose the COL3 domain by skipping of exon 3, which was confirmed by in vitro analysis. The patients were of normal height and had minimal complaints with phenotypes being more severe in male patients. The radiographic phenotypes of the patients were relatively milder than those of previously reported cases, and were indistinguishable to common, idiopathic OA. © 2004 Wiley-Liss, Inc.
Co-reporter:Eiji Nakashima;Akihiko Mabuchi;Mitsuru Kubota;Satoshi Ishikiriyama;Hirofumi Ohashi;Gen Nishimura
American Journal of Medical Genetics Part A 2005 Volume 132A(Issue 1) pp:
Publication Date(Web):2 NOV 2004
DOI:10.1002/ajmg.a.30348
Co-reporter:Akihiko Mabuchi;Shigeki Momohara;Hirofumi Ohashi;Yoshio Takatori;Nobuhiko Haga;Gen Nishimura
American Journal of Medical Genetics Part A 2004 Volume 129A(Issue 1) pp:
Publication Date(Web):7 JUN 2004
DOI:10.1002/ajmg.a.30164
Mutations in the gene encoding cartilage oligomeric matrix protein (COMP) cause two common skeletal dysplasias, pseudoachondroplasia (PSACH) and multiple epiphyseal dysplasia (MED). At present, diagnosis of these diseases is based primarily on clinical and radiographic findings and is sometimes erroneous, particularly in adult patients. However, genetic diagnosis is difficult, because COMP mutations are scattered throughout the gene and five additional disease genes for MED exist. There is evidence that circulating COMP may serve as a molecular indicator of a variety of diseases affecting cartilage. Therefore, we investigated plasma COMP concentrations in 21 patients with PSACH or MED. Of these, six PSACH and seven MED patients carried COMP mutations, and the remaining eight MED patients lacked mutations in COMP. We observed significantly decreased plasma COMP levels in patients with COMP mutations compared with controls (P < 0.0001). In addition, plasma COMP levels were significantly decreased in MED patients carrying mutations in COMP relative to those who lacked COMP mutations (P = 0.001). Our results indicate that circulating COMP levels reflect genetic abnormalities in COMP, providing an easier, more rapid and cost-efficient method for diagnosing PSACH and particularly for MED. © 2004 Wiley-Liss, Inc.
Co-reporter:Eiji Nakashima;Akihiko Mabuchi;Kenichi Kashimada;Toshikazu Onishi;Junwei Zhang;Hirofumi Ohashi;Gen Nishimura
American Journal of Medical Genetics Part A 2003 Volume 123A(Issue 3) pp:
Publication Date(Web):13 MAY 2003
DOI:10.1002/ajmg.a.20281
We examined 12 Japanese patients with metaphyseal chondrodysplasia (MCD) for mutations in the ribonuclease mitochondrial RNA processing gene (RMRP), and identified four novel mutations in two patients with typical and atypical cartilage-hair hypoplasia (CHH), a form of MCD characterized by extra-skeletal manifestations including hypoplastic hair and defective immunity. A patient with typical CHH had a 17-bp duplication at +3 and a de novo 182G > A. The other patient with atypical CHH had a 17-bp insertion at −20 and a 218A > G. Expression analysis revealed that the allele with this insertion mutation in the promoter region silenced the gene. Spectrum analysis of the mutations and polymorphisms in RMRP showed marked difference between the Japanese and other ethnic groups. Such ethnic and phenotypic difference should be taken into account in mutation analysis of the gene. © 2003 Wiley-Liss, Inc.
Co-reporter:Juntaro Matsuyama, Akihiko Mabuchi, Junwei Zhang, Aritoshi Iida, Toshiyuki Ikeda, Mamori Kimizuka and Shiro Ikegawa
Journal of Human Genetics 2003 48(4) pp:173-176
Publication Date(Web):March 11, 2003
DOI:10.1007/s10038-003-0003-9
Tibial hemimelia is a rare congenital anomaly characterized by deficiency of the tibia with relatively intact fibula. Tibial hemimelia is identified as a solitary disorder, or a part of more complex malformation syndromes. Although the majority of cases with tibial hemimelia are sporadic, affected families with possible autosomal dominant or autosomal recessive inheritance have been reported. Here we report a pair of sibs, 6- and 2-year-old Japanese boys, with tibial hemimelia born to unrelated, phenotypically normal parents. The type of tibial hemimelia and associated malformations of hands and feet was quite different between the brothers. The elder brother was compatible with the Gollop-Wolfgang complex, and the younger brother with tibial agenesis-ectrodactyly syndrome. Screening of mutation by direct sequencing of candidate genes including Sonic hedgehog, HOXD-11, and HOXD-12 was unable to identify a disease-causing mutation.
Co-reporter:Toshiyuki Ikeda, Akihiko Mabuchi, Akira Fukuda, Hisatada Hiraoka, Akira Kawakami, Seizo Yamamoto, Hideto Machida, Yoshio Takatori, Hiroshi Kawaguchi, Kozo Nakamura and Shiro Ikegawa
Journal of Human Genetics 2001 46(9) pp:538-543
Publication Date(Web):
DOI:10.1007/s100380170036
Osteoarthritis (OA) is one of the most common musculoskeletal disorders and is characterized by degeneration of articular cartilage. Sulfation of extracellular matrix proteins in articular cartilage is an important step in maintaining normal cartilage metabolism. Two sulfation-related genes have been reported as the causal genes of severe chondrodysplasias: mutations in PAPSS2 (3'-phosphoadenosine 5'-phosphosulfate synthase 2) cause spondylo-epimetaphyseal dysplasia (SEMD), and mutations in SLC26A2 (solute carrier family 26, member 2) cause diastrophic dysplasia. Given their critical roles in cartilage metabolism and the severe phenotypes that result from mutations in these genes, we examined PAPSS2 and SLC26A2 as candidate susceptibility loci for OA. We identified sequence polymorphisms in the coding and core promoter regions of these genes and analyzed their potential association with knee OA within the Japanese population. Ten sequence polymorphisms were detected in PAPSS2 and five in SLC26A2. An association analysis showed suggestive association of one minor polymorphism in the promoter region of SLC26A2. This 4-bp adenine deletion allele, del4A, was over-represented in knee OA (P = 0.043, odds ratio = 3.43) and is thought to confer a minor susceptibility to knee OA within the Japanese population. Haplotype analysis showed no evidence of association with the two genes, however, excluding them as major susceptibility loci for knee OA.
Co-reporter:Akihiko Mabuchi;Nobuhiko Haga;Toshiyuki Ikeda;Noriyo Manabe;Hirofumi Ohashi;Yoshio Takatori;Kozo Nakamura
American Journal of Medical Genetics Part A 2001 Volume 104(Issue 2) pp:
Publication Date(Web):8 OCT 2001
DOI:10.1002/ajmg.10067
Pseudoachondroplasia (PSACH) is a common skeletal dysplasia characterized by disproportionate short stature, early-onset osteoarthrosis, and dysplasia of the spine, epiphysis, and metaphysis. Multiple epiphyseal dysplasia (MED) is a similar but less severe disorder characterized by dysplasia of the epiphysis. Both disorders are caused by mutations in the cartilage oligomeric matrix protein (COMP) gene. COMP mutations cluster in a region of the gene that encodes calmodulin-like repeats (CLRs) and correlate closely with disease severity. Typically, mutations in exon 13 that composes the seventh CLR produce severe PSACH phenotypes, whereas mutations found elsewhere in the gene produce mild PSACH or MED phenotypes. We have identified a PSACH patient carrying a novel mutation in exon 18 of COMP that composes the C-terminal globular domain. This mutation produced a severe PSACH phenotype with marked short stature and deformities of the spine and extremities. Our results extend the range of disease-causing mutations within the COMP gene and demonstrate the importance of the additional domain of COMP protein in its in vivo function. © 2001 Wiley-Liss, Inc.
Co-reporter:Akihiko Mabuchi, Toshiyuki Ikeda, Akira Fukuda, Yu Koshizuka, Hisatada Hiraoka, Kota Miyoshi, Nobuhiko Haga, Hiroshi Kawaguchi, Akira Kawakami, Seizo Yamamoto, Yoshio Takatori, Kozo Nakamura and Shiro Ikegawa
Journal of Human Genetics 2001 46(8) pp:456-462
Publication Date(Web):
DOI:10.1007/s100380170045
Osteoarthrosis (OA) is a common cause of musculoskeletal disability characterized by late-onset degeneration of articular cartilage. Although several candidate genes have been reported, susceptibility genes for OA remain to be determined. Hereditary osteochondral dysplasias produce severe, early-onset OA and hence are models for common idiopathic OA. Among them are pseudoachondroplasia and multiple epiphyseal dysplasia, both of which are caused by mutations in the cartilage oligomeric matrix protein (COMP) gene. Therefore, COMP may be a susceptibility gene for OA. We screened for polymorphisms by direct sequencing of all exons of the COMP gene with their flanking intron sequences and the promoter region. We identified 16 polymorphisms, of which 12 were novel. Using six polymorphisms spanning the entire COMP gene, we examined the association of COMP in Japanese patients with OA of the knee and hip joints. Genotype and allele frequencies of the polymorphisms were not significantly different between OA and control groups, and there was no significant difference in haplotypes. These results do not support an association between COMP and OA in the Japanese population.
Co-reporter:Shiro Ikegawa;Hirofumi Ohashi;Tsutomu Ogata;Akira Honda;Masato Tsukahara;Toshihide Kubo;Mamori Kimizuka;Masanori Shimode;Tomonobu Hasegawa;Gen Nishimura;Yusuke Nakamura
American Journal of Medical Genetics Part A 2000 Volume 94(Issue 4) pp:
Publication Date(Web):13 OCT 2000
DOI:10.1002/1096-8628(20001002)94:4<300::AID-AJMG7>3.0.CO;2-3
Chondrodysplasia punctata (CDP) is a heterogeneous group of skeletal dysplasias characterized by stippled epiphyses. A subtype of CDP, X-linked dominant chondrodysplasia punctata (CDPX2), known also as Conradi-Hünermann-Happle syndrome, is a rare skeletal dysplasia characterized by short stature, craniofacial defects, cataracts, ichthyosis, coarse hair, and alopecia. The cause of CDPX2 was unknown until recent identification of mutations in the gene encoding Δ8,Δ7 sterol isomerase emopamil-binding protein (EBP). Twelve different EBP mutations have been reported in 14 patients with CDPX2 or unclassified CDP, but with no evidence of correlation between phenotype and nature of the mutation. To characterize additional mutations and investigate possible phenotype-genotype correlation, we sequenced the entire EBP gene in 8 Japanese individuals with CDP; 5 of them presented with a CDPX2 phenotypes. We found EBP mutations in all 5 CDPX2 individuals, but none in non-CDPX2 individuals. Three of these CDPX2 individuals carried novel nonsense mutations in EBPand the other two, separate missense mutations that had been reported also in different ethnic groups. Our results, combined with previous information, suggest all EBP mutations that produce truncated proteins result in typical CDPX2, whereas the phenotypes resulted from missense mutations are not always typical for CDPX2. Patients with nonsense mutations showed abnormal sterol profiles consistent with a defect in Δ8,Δ7 sterol isomerase. X-inactivation patterns of the patients showed no skewing, an observation that supports the assumption that inactivation of the EBP gene occurs at random in affected individuals. Am. J. Med. Genet. 94:300–305, 2000. © 2000 Wiley-Liss, Inc.
Co-reporter:Shiro Ikegawa, Minoru Isomura, Yu Koshizuka and Yusuke Nakamura
Journal of Human Genetics 1999 44(5) pp:337-342
Publication Date(Web):
DOI:10.1007/s100380050172
Large-scale DNA sequencing, coupled with in silico gene trapping, is a robust approach to identifying unknown genes in selected genomic regions. Using this approach we have isolated a novel human gene, PROSC (for proline synthetase co-transcribed [bacterial homolog]), from human chromosome 8p11.2, and its mouse counterpart. The human PROSC gene spanned 17kb of genomic DNA; its cDNA was 2530bp long, with 8 exons that included an open reading frame of 825bp (275 amino acids). The mouse cDNA (Prosc), 1995bp long, was predicted to encode 274 amino acids. PROSC is ubiquitously expressed in human tissues and has been highly conserved among divergent species from bacteria to mammals, suggesting its important cellular function. The gene product is likely to be a soluble cytoplasmic protein, but its function remains to be determined.
Co-reporter:Aritoshi Iida, Naoya Hosono, Motoki Sano, Tetsumasa Kamei, Shuichi Oshima, Torao Tokuda, Masahiro Nakajima, Michiaki Kubo, Yusuke Nakamura, Shiro Ikegawa
Neurobiology of Aging (August 2012) Volume 33(Issue 8) pp:1843.e19-1843.e24
Publication Date(Web):1 August 2012
DOI:10.1016/j.neurobiolaging.2011.12.037
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by selective motor neuron death in the brain and spinal cord. Many disease genes for ALS have been identified; however, each disease gene is responsible for very small fractions of ALS. Recently, mutations of the gene encoding optineurin (OPTN) are reported in familial and sporadic ALS. OPTN is also responsible for a small number of ALS, 3.8% of familial and 0.29% of sporadic ALS in Japanese. The low prevalence may be an underestimation due to incomplete screening of the mutation. To examine OPTN mutations more extensively, we screened the OPTN deletions using a quantitative PCR system. We examined 710 Japanese ALS subjects who had previously been found to have no OPTN mutations by a screening using a PCR-direct sequence strategy. We identified 3 kinds of deletions in 5 patients; one was homozygous, and the remaining were heterozygous. All deletions occurred due to the Alu-mediated recombination and are expected to result in null alleles. Our results suggest that the OPTN deletion mutation in ALS is not infrequent and the prevalence of the OPTN mutation in Japanese sporadic ALS is considerably high.
Co-reporter:Aritoshi Iida, Tetsumasa Kamei, Motoki Sano, Shuichi Oshima, Torao Tokuda, Yusuke Nakamura, Shiro Ikegawa
Neurobiology of Aging (April 2012) Volume 33(Issue 4) pp:786-790
Publication Date(Web):1 April 2012
DOI:10.1016/j.neurobiolaging.2010.06.017
Mutations in TARDBP encoding TDP (TAR DNA binding protein)-43 have been reported in familial and sporadic amyotrophic lateral sclerosis (ALS), but mostly in Caucasians. In other ethnic groups, four types of mutations are found in familial ALS. In sporadic ALS, the TARDBP mutations frequency is low in Caucasians (0–5%) and no mutation has been found in other ethnic groups. To examine spectrum of TARDBP mutations and its frequency in Japanese, we screened the TARDBP mutation in 721 Japanese ALS by direct sequencing. We identified a novel mutation, c.1069G > A (p.Gly357Ser) and a known mutation in sporadic ALS. One patient was homozygous for p.Gly357Ser, which was the first for TARDBP mutation. Our study showed that TARDBP mutations also occur in non-Caucasian sporadic ALS. The estimated frequency of the TARDBP mutation in sporadic ALS is 0.29% in Japanese. The mutation frequency in familial ALS in Japanese is also similar to that in Caucasian, and is ∼10 times higher than that in Japanese sporadic ALS.
Co-reporter:Masahiro Nakajima, Ikuyo Kou, Hirofumi Ohashi, Genetic Study Group of the Investigation Committee on the Ossification of Spinal Ligaments, Shiro Ikegawa
The American Journal of Human Genetics (7 July 2016) Volume 99(Issue 1) pp:
Publication Date(Web):7 July 2016
DOI:10.1016/j.ajhg.2016.05.018
Ossification of the posterior longitudinal ligament of the spine (OPLL) is a common spinal disorder that results from ectopic ossification of the posterior longitudinal ligament and causes intractable myelopathy and radiculopathy. In a previous genome-wide association study (GWAS), we found six loci associated with OPLL; however, susceptibility genes in these loci have not been identified yet. Here, we examined one of the GWAS loci and identified RSPO2 (encoding R-spondin 2) as a susceptibility gene for OPLL. R-spondin 2 is a secreted agonist of canonical Wnt-β-catenin signaling. RSPO2 was decreased in the early stage of chondrocyte differentiation. R-spondin 2 inhibited expression of genes encoding early chondrocyte differentiation markers by activating Wnt-β-catenin signaling. rs374810, the most significantly associated SNP in the GWAS locus in chromosomal region 8q23.1 was located in the chondrocyte promoter region of RSPO2. A transcription factor, CCAAT-enhancer-binding protein β (C/EBPβ), specifically bound to the RSPO2 core promoter region containing rs374810 and increased RSPO2 expression. The risk allele of rs374810 affected the binding of the promoter with C/EBPβ and decreased the RSPO2 transcription in vitro and in vivo. Our genetic and functional data indicate that RSPO2 is a susceptibility gene for OPLL.
Co-reporter:Masahiro Nakajima, Shuji Mizumoto, Noriko Miyake, Ryo Kogawa, Aritoshi Iida, Hironori Ito, Hiroshi Kitoh, Aya Hirayama, Hiroshi Mitsubuchi, Osamu Miyazaki, Rika Kosaki, Reiko Horikawa, Angeline Lai, Roberto Mendoza-Londono, Lucie Dupuis, David Chitayat, Andrew Howard, Gabriela F. Leal, Denise Cavalcanti, Yoshinori Tsurusaki, Hirotomo Saitsu, et al.
The American Journal of Human Genetics (6 June 2013) Volume 92(Issue 6) pp:
Publication Date(Web):6 June 2013
DOI:10.1016/j.ajhg.2013.04.003
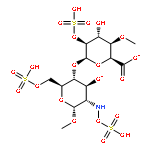
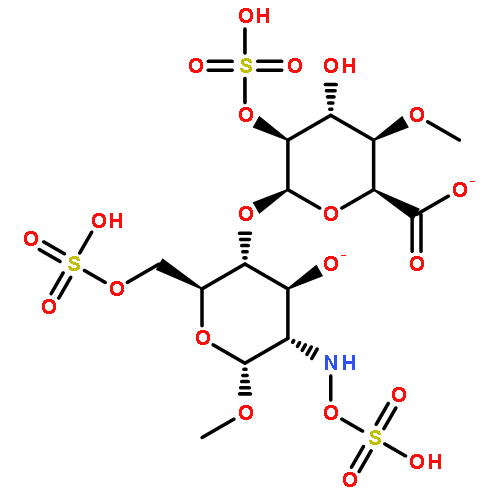
Proteoglycans (PGs) are a major component of the extracellular matrix in many tissues and function as structural and regulatory molecules. PGs are composed of core proteins and glycosaminoglycan (GAG) side chains. The biosynthesis of GAGs starts with the linker region that consists of four sugar residues and is followed by repeating disaccharide units. By exome sequencing, we found that B3GALT6 encoding an enzyme involved in the biosynthesis of the GAG linker region is responsible for a severe skeletal dysplasia, spondyloepimetaphyseal dysplasia with joint laxity type 1 (SEMD-JL1). B3GALT6 loss-of-function mutations were found in individuals with SEMD-JL1 from seven families. In a subsequent candidate gene study based on the phenotypic similarity, we found that B3GALT6 is also responsible for a connective tissue disease, Ehlers-Danlos syndrome (progeroid form). Recessive loss-of-function mutations in B3GALT6 result in a spectrum of disorders affecting a broad range of skeletal and connective tissues characterized by lax skin, muscle hypotonia, joint dislocation, and spinal deformity. The pleiotropic phenotypes of the disorders indicate that B3GALT6 plays a critical role in a wide range of biological processes in various tissues, including skin, bone, cartilage, tendon, and ligament.
Co-reporter:Yoji Ogura, Ikuyo Kou, Shigenori Miura, Atsushi Takahashi, Leilei Xu, Kazuki Takeda, Yohei Takahashi, Katsuki Kono, Noriaki Kawakami, Koki Uno, Manabu Ito, Shohei Minami, Ikuho Yonezawa, Haruhisa Yanagida, Hiroshi Taneichi, Zezhang Zhu, Taichi Tsuji, Teppei Suzuki, Hideki Sudo, Toshiaki Kotani, Kota Watanabe, et al.
The American Journal of Human Genetics (6 August 2015) Volume 97(Issue 2) pp:
Publication Date(Web):6 August 2015
DOI:10.1016/j.ajhg.2015.06.012
Adolescent idiopathic scoliosis (AIS) is the most common spinal deformity. We previously conducted a genome-wide association study (GWAS) and detected two loci associated with AIS. To identify additional loci, we extended our GWAS by increasing the number of cohorts (2,109 affected subjects and 11,140 control subjects in total) and conducting a whole-genome imputation. Through the extended GWAS and replication studies using independent Japanese and Chinese populations, we identified a susceptibility locus on chromosome 9p22.2 (p = 2.46 × 10−13; odds ratio = 1.21). The most significantly associated SNPs were in intron 3 of BNC2, which encodes a zinc finger transcription factor, basonuclin-2. Expression quantitative trait loci data suggested that the associated SNPs have the potential to regulate the BNC2 transcriptional activity and that the susceptibility alleles increase BNC2 expression. We identified a functional SNP, rs10738445 in BNC2, whose susceptibility allele showed both higher binding to a transcription factor, YY1 (yin and yang 1), and higher BNC2 enhancer activity than the non-susceptibility allele. BNC2 overexpression produced body curvature in developing zebrafish in a gene-dosage-dependent manner. Our results suggest that increased BNC2 expression is implicated in the etiology of AIS.