Co-reporter:Hans J. Reich and Robert J. Hondal
ACS Chemical Biology 2016 Volume 11(Issue 4) pp:821
Publication Date(Web):March 7, 2016
DOI:10.1021/acschembio.6b00031
The authors were asked by the Editors of ACS Chemical Biology to write an article titled “Why Nature Chose Selenium” for the occasion of the upcoming bicentennial of the discovery of selenium by the Swedish chemist Jöns Jacob Berzelius in 1817 and styled after the famous work of Frank Westheimer on the biological chemistry of phosphate [Westheimer, F. H. (1987) Why Nature Chose Phosphates, Science 235, 1173–1178]. This work gives a history of the important discoveries of the biological processes that selenium participates in, and a point-by-point comparison of the chemistry of selenium with the atom it replaces in biology, sulfur. This analysis shows that redox chemistry is the largest chemical difference between the two chalcogens. This difference is very large for both one-electron and two-electron redox reactions. Much of this difference is due to the inability of selenium to form π bonds of all types. The outer valence electrons of selenium are also more loosely held than those of sulfur. As a result, selenium is a better nucleophile and will react with reactive oxygen species faster than sulfur, but the resulting lack of π-bond character in the Se–O bond means that the Se-oxide can be much more readily reduced in comparison to S-oxides. The combination of these properties means that replacement of sulfur with selenium in nature results in a selenium-containing biomolecule that resists permanent oxidation. Multiple examples of this gain of function behavior from the literature are discussed.
Co-reporter:Kristin N. Plessel, Amanda C. Jones, Daniel J. Wherritt, Rebecca M. Maksymowicz, EricT. Poweleit, and Hans J. Reich
Organic Letters 2015 Volume 17(Issue 10) pp:2310-2313
Publication Date(Web):April 27, 2015
DOI:10.1021/acs.orglett.5b00650
Unexpectedly high rates of reaction between alkyllithium reagents and amides, compared to esters and ketones, were observed by Rapid Inject NMR and competition experiments. Spectroscopic investigations with 4-fluorophenyllithium (ArLi, mixture of monomer and dimer in THF) and a benzoate ester identified two reactive intermediates, a homodimer of the tetrahedral intermediate, stable below −100 °C, and a mixed dimer with ArLi. Direct formation of dimers suggested that the ArLi dimer may be the reactive aggregate rather than the usually more reactive monomer. In contrast, RINMR experiments with ketones demonstrated that the ArLi monomer was the reactive species.
Co-reporter:Hans J. Reich
Chemical Reviews 2013 Volume 113(Issue 9) pp:7130
Publication Date(Web):August 13, 2013
DOI:10.1021/cr400187u
Co-reporter:Hans J. Reich
The Journal of Organic Chemistry 2012 Volume 77(Issue 13) pp:5471-5491
Publication Date(Web):May 17, 2012
DOI:10.1021/jo3005155
This Perspective describes a series of research projects that led the author from an interest in lithium reagents as synthetically valuable building blocks to studies aimed at understanding the science behind the empirical art developed by synthetic chemists trying to impose their will on these reactive species. Understanding lithium reagent behavior is not an easy task; since many are mixtures of aggregates, various solvates are present, and frequently new mixed aggregates are formed during their reactions with electrophiles. All of these species are typically in fast exchange at temperatures above −78 °C. Described are multinuclear NMR experiments at very low temperatures aimed at defining solution structures and dynamics and some kinetic studies, both using classic techniques as well as the rapid inject NMR (RINMR) technique, which can in favorable cases operate on multispecies solutions without the masking effect of the Curtin–Hammett principle.
Co-reporter:Kristopher J. Kolonko ; Daniel J. Wherritt
Journal of the American Chemical Society 2011 Volume 133(Issue 42) pp:16774-16777
Publication Date(Web):September 22, 2011
DOI:10.1021/ja207218f
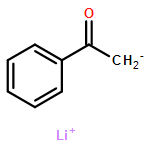
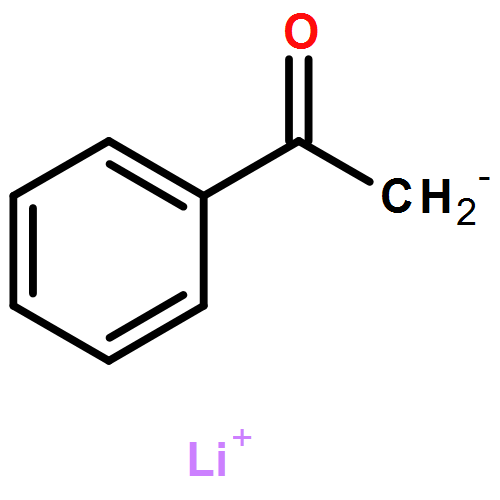
Lithium enolates are widely used nucleophiles with a complicated and only partially understood solution chemistry. Deprotonation of 4-fluoroacetophenone in THF with lithium diisopropylamide occurs through direct reaction of the amide dimer to yield a mixed enolate-amide dimer (3), then an enolate homodimer (1-Li)2, and finally an enolate tetramer (1-Li)4, the equilibrium structure. Aldol reactions of both the metastable dimer and the stable tetramer of the enolate were investigated. Each reacted directly with the aldehyde to give a mixed enolate-aldolate aggregate, with the dimer only about 20 times as reactive as the tetramer at −120 °C.
Co-reporter:Kristopher J. Kolonko ; Margaret M. Biddle ; Ilia A. Guzei
Journal of the American Chemical Society 2009 Volume 131(Issue 32) pp:11525-11534
Publication Date(Web):July 27, 2009
DOI:10.1021/ja903479p
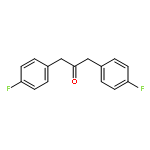
Multinuclear NMR spectroscopic studies at low temperature (−110 to −150 °C) revealed that lithium p-fluorophenolate and the lithium enolates of cyclohexanone, cyclopentanone and 4-fluoroacetophenone have tetrameric structures in THF/Et2O and THF/Et2O−HMPA by study of the effects of the addition of HMPA. The Z and E isomers of the lithium enolate of 1,3-bis-(4-fluorophenyl)-2-propanone (5F-Li) show divergent behavior. The Z isomer is completely dimeric in pure diethyl ether, and mostly dimeric in 3:2 THF/ether, where monomer could be detected in small amounts. TMTAN and PMDTA convert Z-5F-Li to a monomeric amine complex, and HMPA converts it partially to monomers, and partially to lithiate species (RO)2Li− and (RO)3Li2−. Better characterized solutions of these lithiates were prepared by addition of phosphazenium enolates (using P4-tBu base) to the lithium enolate in 1:1 ratio to form triple ion (RO)2Li− P4H+, or 2:1 ratio to form the higher lithiate (RO)3Li2− (P4H+)2) (quadruple ions). The E isomer of 5F-Li is also dimeric in 3:2 THF/Et2O solution, but is not detectably converted to monomer either by PMDTA or HMPA. In contrast to Z-5F-Li, the E isomer is tetrameric in diethyl ether even in the presence of excess HMPA. Thus for the two isomers of 5F six different enolate structures were characterized: tetramer, dimer, CIP-monomer, SIP-monomer, triple ion, and quadruple ion.
Co-reporter:Hans J. Reich Dr.;Martin J. Bevan;Birgir Ö. Gudmundsson Dr.;Craig L. Puckett Dr.
Angewandte Chemie 2002 Volume 114(Issue 18) pp:
Publication Date(Web):13 SEP 2002
DOI:10.1002/1521-3757(20020916)114:18<3586::AID-ANGE3586>3.0.CO;2-U
Einen Li/Te-Austausch über den at-Komplex ohne Beteiligung des direkten Austauschs (d. h. kd=0) ergaben NMR-Studien an einer intermolekularen Reaktion (siehe Schema; M=Te, Ar=5-Diphenylphosphanyl-2-thienyl). Dagegen wird bei einem intramolekularen Li/Se-Austausch der nachweisbare at-Komplex durch den direkten Austausch umgangen (kd≫kat).





![Lithium, [(4-fluorophenyl)ethynyl]-](http://img.cochemist.com/ccimg/62500/62440-16-8.png)
![Lithium, [(4-fluorophenyl)ethynyl]-](http://img.cochemist.com/ccimg/62500/62440-16-8_b.png)


![4,7,13,18-tetraoxa-1,10-diazabicyclo[8.5.5]icosane](http://img.cochemist.com/ccimg/31300/31250-06-3.png)
![4,7,13,18-tetraoxa-1,10-diazabicyclo[8.5.5]icosane](http://img.cochemist.com/ccimg/31300/31250-06-3_b.png)