Co-reporter:Dashuai Wang, Yu Gao, Yanhui Liu, Di Jin, Yury Gogotsi, Xing Meng, Fei Du, Gang Chen, and Yingjin Wei
The Journal of Physical Chemistry C June 22, 2017 Volume 121(Issue 24) pp:13025-13025
Publication Date(Web):June 5, 2017
DOI:10.1021/acs.jpcc.7b03057
The potential of a Ti2N monolayer and its Ti2NT2 derivatives (T = O, F, and OH) as anode materials for lithium-ion and beyond-lithium-ion batteries has been investigated by the first-principles calculations. The bare and terminated monolayers are metallic compounds with high electronic conductivity. The diffusion barriers on bare Ti2N monolayer are predicted to be 21.5 meV for Li+, 14.0 meV for Na+, 7.0 meV for K+, 75.9 meV for Mg2+, and 38.0 meV for Ca2+, which are the lowest values reported for state-of-the-art two-dimensional energy storage materials. The functional groups on Ti2NT2 increase the diffusion barriers by about 1 order of magnitude. The calculated capacities for the monovalent cations on Ti2N and Ti2NT2 are close to that of the conventional graphite anode in lithium-ion batteries. In comparison, the capacities for Mg2+ on Ti2N and Ti2NT2 are more than 2000 mAh g–1 due to the two-electron reaction and multilayer adsorption of Mg2+. Comparison of the electrochemical performances of Ti2N and Ti2C suggests that Ti2N is a more promising anode material than Ti2C due to its lower diffusion barriers for various cations.
Co-reporter:Dashuai Wang;Yanhui Liu;Xing Meng;Yingjin Wei;Yingying Zhao;Qiang Pang
Journal of Materials Chemistry A 2017 vol. 5(Issue 40) pp:21370-21377
Publication Date(Web):2017/10/17
DOI:10.1039/C7TA06944H
First-principles calculations based on density functional theory were carried out to investigate the electrochemical performance of monolayer VS2 for Li-, K-, Mg- and Al-ion batteries. A VS2 monolayer shows differential storage ability for various cations, able to adsorb three layers of Li, two layers of Mg, one layer of K, and 1/9 layer of Al on both sides of the monolayer, producing theoretical capacities of 1397, 1863, 466, and 78 mA h g−1 for Li, Mg, K, and Al, respectively. The average working voltages of VS2 monolayers for Li+, K+ and Mg2+ are close to those of metallic Li, K, and Mg, suggesting that they can be used as anode materials in these rechargeable batteries. The adsorbed cations form a honeycomb-stacking lattice on VS2 monolayers, similar to the plating process of Li, K, and Mg metal anodes. More interestingly, the honeycomb Li lattice is different from the body-centered cubic lattice of a Li metal anode, which provides very small diffusion barriers, resulting in the high rate capability of VS2 monolayer in Li-ion batteries.
Co-reporter:Dashuai Wang;Yu Gao;Yanhui Liu;Yury Gogotsi;Xing Meng;Yingjin Wei
Journal of Materials Chemistry A 2017 vol. 5(Issue 47) pp:24720-24727
Publication Date(Web):2017/12/05
DOI:10.1039/C7TA09057A
Chloride ion adsorption on Ti2C monolayers was theoretically investigated. Electrochemical parameters, including specific capacity, chloride ion (Cl−) diffusion barrier, and discharge voltage profile, were studied via first-principles calculations. The most favorable Cl− adsorption configuration was identified using a partial particle swarm optimization algorithm and the results showed that Cl− adsorption onto Ti2C monolayers achieved a large theoretical capacity (331 mA h g−1), high working voltage (4.0–3.5 V), and low diffusion barrier (0.22 eV). This resulted in excellent rate capability and a large specific energy of 1269 W h kg−1 at the material level. The effects of terminal O, F, and OH groups on Cl− adsorption onto Ti2C monolayer were also studied, which showed that Cl− could not be adsorbed onto O and F terminated Ti2C monolayers. In comparison, Cl− adsorption onto OH terminated Ti2C was allowed but generated a smaller specific capacity (126 mA h g−1) and lower working voltage (3.5–1.5 V) than a bare Ti2C monolayer.
Co-reporter:Xing Ming, Qiheng Hu, Fang Hu, Fei Du, Yingjin Wei, Gang Chen
Computational Materials Science 2016 Volume 119() pp:33-40
Publication Date(Web):15 June 2016
DOI:10.1016/j.commatsci.2016.03.030
The crystal structure, ferroelectric polarization, magnetism, and electronic structure in Fe-substituted tetragonal BiCoO3 at concentrations of 25% and 50% are investigated within the framework of density functional theory. The C-type antiferromagnetic (AFM) spin configurations with intra-layer AFM coupling interactions are energetically favorable among all the considered magnetic ordering states. Fe substitutions produce 1 μB net magnetic moment in Fe-substituted BiCoO3 system. Using the point charge model, the ferroelectric polarizations are predicted to be as high as 165 and 163 μC/cm2 for 25% and 50% Fe substitution. Electronic band structures reveal that the Fe-substituted BiCoO3 systems are ferrimagnetic insulator. Present first-principles calculation results demonstrate that Fe substitutional doping may produce multiferroic materials simultaneously showing ferrimagnetic and excellent ferroelectric properties.
Co-reporter:Yongmao Cai, Zu-Fei Huang, Xing Meng, Xing Ming, Chunzhong Wang, Gang Chen
Solid State Sciences 2011 Volume 13(Issue 2) pp:350-355
Publication Date(Web):February 2011
DOI:10.1016/j.solidstatesciences.2010.11.034
Transformations of the four polymorphs and magnetic transition of BaRuO3 under high pressure have been studied using first-principles calculations. We search for the low-enthalpy structures by relaxing four initial structural configurations under high pressure. We find that BaRuO3 exhibits sequential structural changes from nine-layer rhombohedral (9R), four-layer hexagonal (4H), six-layer hexagonal (6H), and finally to cubic perovskite (3C) structure, consistent well with the experiments. The three hexagonal crystallographic forms (9R, 4H and 6H) of BaRuO3 show paramagnetic states, whereas 3C–BaRuO3 displays a ferromagnetic ground state. Along with the pressure increasing, the magnetic moment of Ru in the 3C–BaRuO3 first come through a discontinuous drop around the critical pressure 26 GPa, then becomes smaller gradually until zero at 180 GPa. According to the theory of Moruzzi, this magnetic–nonmagnetic transition belongs to the type-III transition.The calculated pressure dependence of the enthalpies (a) and the volumes (b) of BaRuO3, reveals the pressure-induced phase transformation of BaRuO3.
Co-reporter:Yongmao Cai, Yingjin Wei, Xing Ming, Fei Du, Xing Meng, Chunzhong Wang, Gang Chen
Solid State Communications 2011 Volume 151(Issue 10) pp:798-801
Publication Date(Web):May 2011
DOI:10.1016/j.ssc.2011.02.031
The phase transition in the perovskite (Pv) SrRuO3 under pressure has been studied by using first-principles calculations based on density functional theory. The post-perovskite (Ppv) phase transition of SrRuO3 will take place under hydrostatic pressure of about 40 GPa. The stability of Ppv- SrRuO3 is justified by the enthalpy calculations, and this phase transition accompanies volume disconnection and magnetic moment collapses. The crystallographic data and the electronic structure of Ppv- SrRuO3 are also predicted. The crystal structure of Ppv- SrRuO3 is similar to that of Ppv- CaRuO3. A non-magnetic ground state is found in Ppv- SrRuO3. The strong hybridization of Ru and O is evident in the electronic structures of both phases. We expect that these results will help further understanding of SrRuO3 under high pressure.Highlights► The post-perovskite (Ppv) phase transition of SrRuO3 has been predicted. ► The Ppv phase transition accompanies volume disconnection and magnetic moment collapses. ► The crystallographic data and the electronic structure of Ppv- SrRuO3 have been revealed.
Co-reporter:Yongmao Cai, Zu-Fei Huang, Xing Ming, Chunzhong Wang, Gang Chen
Journal of Alloys and Compounds 2010 Volume 505(Issue 2) pp:L23-L26
Publication Date(Web):3 September 2010
DOI:10.1016/j.jallcom.2010.06.130
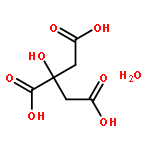
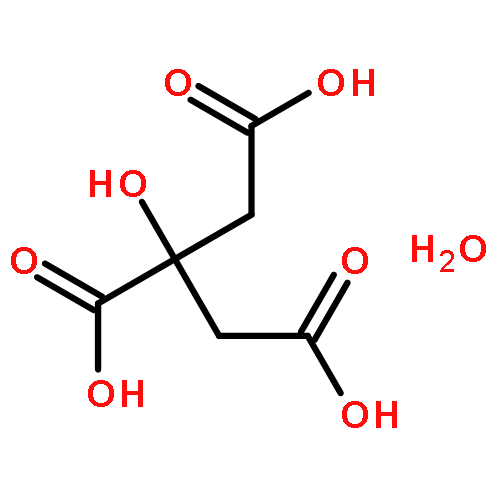
The charge disproportionation of V ions in the low-temperature rhombohedral phase of AlV2O4 has been studied by first-principles density functional theory (DFT) computations. The calculation results show that there exists an alone V ion (V1) and a “heptamer” constructed by two types of V ions (one V2 and six V3) in the rhombohedral AlV2O4. The theoretical results are consistent with previous experimental reports (Horibe et al., Phys. Rev. Lett. 96 (2006) 086406; Phys. Rev. Lett. 96 (2006) 169901). The valence states of the three types of V ions are found to be +(2.5 − δ1), +(2.5 + δ2) and +(2.5 + (δ1 − δ2)/6) (δ1 > δ2 > 0), respectively. The three types of V ions with different charges arrange periodically layer by layer along the c-axis direction with a sequence as V1–V3–V2–V3–V1.
Co-reporter:X. Li, Y.J. Wei, H. Ehrenberg, D.L. Liu, S.Y. Zhan, C.Z. Wang, G. Chen
Journal of Alloys and Compounds 2009 Volume 471(1–2) pp:L26-L28
Publication Date(Web):5 March 2009
DOI:10.1016/j.jallcom.2008.03.129
Inverse spinel LiNiVO4 was prepared by a soft-chemical technique. The structural properties of the as-prepared and electrochemically Li-extracted LiNiVO4 were investigated by X-ray diffraction and Raman scattering. X-ray diffraction showed that Li-extraction had little effect on the material's crystal structure. Raman scattering showed softening of the V–O bonds and severer distorted [VO4] tetrahedron with Li-extraction. Li-extraction resulted in a decreased electronic conductivity to the material. The relative intensity of the A1g and F2g bands was suggested to be associated with the Li+ content in LixNiVO4.
Co-reporter:Fei Du, Yingjin Wei, Yan Chen, Fang Hu, Xiaofei Bie, Chunzhong Wang, Gang Chen, Guangtian Zou
Solid State Sciences 2009 Volume 11(Issue 11) pp:1866-1869
Publication Date(Web):November 2009
DOI:10.1016/j.solidstatesciences.2009.08.008
The valence state and magnetic properties of hole-doped LiCuVO4 are investigated. By analyzing the Cu 2p core-level photoemission spectra, the holes are suggested to be introduced into Cu2+ site in the formation of Cu3+ ion. The calculation of effective moment also confirms the presence of nonmagnetic Cu3+ ion which should be responsible for the decrease of high-temperature susceptibility after hole doping. At low temperature, the antiferromagnetic transition at 26 K and 2.3 K disappears due to the enhancement of Curie term. Magnetic hysteresis at 2 K shows that there exists a small ferromagnetic moment of 0.15 emu/g in Li0.9CuVO4.
Co-reporter:Zu-Fei Huang, Han-Zhuang Zhang, Chun-Zhong Wang, Deng-Pan Wang, Xing Meng, Xing Ming, Gang Chen
Solid State Sciences 2009 Volume 11(Issue 1) pp:271-274
Publication Date(Web):January 2009
DOI:10.1016/j.solidstatesciences.2008.06.005
Changes with Li extraction in the crystal structure and electronic structure of monoclinic LiMnO2 have been investigated by first-principle calculations using spin-polarized generalized gradient approximation method. It is found that the Li extraction has changed the oxidation state of Mn ion from 3+ to 4+ and thereby induced a phase transition from the monoclinic structure to rhombohedral symmetry, which opens channels for the migration of Mn ions from their own octahedral sites to the interlayer Li vacancies. It is the migration of Mn ions that responds for the phase transition to a spinel-like structure and the severe capacity loss during the electrochemical cycles of monoclinic LiMnO2.Band structures of monoclinic LiMnO2 (a) and its Li-extracted form (b), which reveals the phase transition mechanism in this material with Li extraction.
Co-reporter:Da-Liang Liu, Fei Du, Ying-Jin Wei, Chun-Zhong Wang, Zu-Fei Huang, Xing Meng, Gang Chen, Yan Chen, Shou-Hua Feng
Materials Letters 2009 Volume 63(Issue 1) pp:133-135
Publication Date(Web):15 January 2009
DOI:10.1016/j.matlet.2008.09.035
Polycrystalline lithium nitridocobaltate Li2.5Co0.5N was prepared by solid-state reaction. Both field-cooled (FC) and zero-field-cooled (ZFC) magnetization were measured, in which irreversibility between the ZFC and FC processes was observed and the freezing temperature (Tf) shifted to low temperature with increasing of the applied field. This indicates lack of long range magnetic order in Li2.5Co0.5N .We studied the effect of frequency on the ac susceptibility. It was found out that there was a peak and the peak position shifting with the frequency. A criterion parameter δ, ΔT'/[T'Δlogv], is calculated to be 0.04(1) based the experimental data. The observed magnetic behavior indicates a spin-glass ground state in Li2.5Co0.5N.
Co-reporter:Daliang Liu, Shiying Zhan, Gang Chen, Wencheng Pan, Chunzhong Wang, Yingjin Wei
Materials Letters 2008 Volume 62(Issue 26) pp:4210-4212
Publication Date(Web):15 October 2008
DOI:10.1016/j.matlet.2008.06.036
Li2.6Co0.4 - xCuxN (x = 0, 0.15) anode materials were prepared by conventional solid state reaction. Between both materials, Li2.6Co0.25Cu0.15N exhibited better capacity retention than that of Li2.6Co0.4N. According to electrochemical impedance spectroscopy, the better cycling behavior of Li2.6Co0.25Cu0.15N has been attributed to the improvement in interfacial compatibility between the electrode and electrolyte interface. A possible explanation to this was given. Li2.6Co0.4 - xCuxN/Cu0.04V2O5 full-cells were assembled to investigate the reliability of Li2.6Co0.4 - xCuxN anode materials in practical applications. The Li2.6Co0.25Cu0.15N/Cu0.04V2O5 cell delivered a specific capacity of 260 mA h g− 1, and a specific energy of 505.7 mW h g− 1, which was much higher than that of C/LiCoO2 lithium ion batteries.
Co-reporter:X. Li, Y.J. Wei, H. Ehrenberg, F. Du, C.Z. Wang, G. Chen
Solid State Ionics 2008 Volume 178(39–40) pp:1969-1974
Publication Date(Web):15 March 2008
DOI:10.1016/j.ssi.2008.01.048
LiNi1/3Mn1/3Co1/3O2 compounds were prepared at 700 (sample-A) and 900 °C (sample-B) by a simple wet-chemical process. X-ray diffraction showed the sample-A had a larger Li/Ni site exchange (9.9%) than that of the sample-B (4.4%). Raman scattering analysis suggests strong structural disorder within the octahedral [MO6] units due to different masses and bonding of the M–O bonds, as well as the Li/Ni site exchange in the crystals. The weakening of the Eg band was correlated with the Li/Ni site exchange. XPS analysis indicates that a lower annealing temperature than 900 °C leads to the formation of oxygen deficient LiNi1/3Mn1/3Co1/3O2 − δ. The electrochemical properties of the materials were investigated by charge–discharge experiments and electrochemical impedance spectroscopy. The drastic capacity fading of the sample-A was attributed to its structural properties. The sample-B showed satisfying cycling performance. But the capacity fading in the potential region of 2.5–4.6 V was larger than that in 2.5–4.4 V, which was attributed to the large charge transfer resistance at potentials above 4.4 V.
Co-reporter:Zu-Fei Huang, Chun-Zhong Wang, Xing Meng, Deng-Pan Wang, Gang Chen
Journal of Solid State Chemistry 2006 Volume 179(Issue 5) pp:1602-1609
Publication Date(Web):May 2006
DOI:10.1016/j.jssc.2006.02.016
Electronic structures of monoclinic LiMnO2 and LiMn0.9375Al0.0625O2 with ferromagnetic (FM) and antiferromagnetic (AF) ordering have been investigated by ab initio calculation within spin-polarized generalized gradient approximation method. An Al-doping induced complicated AF configuration has been calculated to be the ground state, which suggests a robust Al-doping effect on the magnetic and electronic structures of the monoclinic LiMnO2. The calculated Mulliken population analyses and partial density of states of Mn-3d and O-2p reveal that a single Al dopant stabilizes its six nearest-neighbor Mn ions in their respective octahedral sites, thereby hindering the migration of Mn ions into the interlayer Li sites during the Li intercalation–deintercalation and therefore improving both the structural stability and the electrochemical performance of the material. Additionally, it is found out that the Al-doping can decrease the JT effect and increase the intercalation voltage. The Al-doping-induced negative formation energy indicates that 6.25% Mn ions in monoclinic LiMnO2 can be substituted stably by Al ions, to which the equilibrium but not metastable phase of monoclinic LiMn0.9375Al0.0625O2 can be attributed.Total density of states (DOS) around EF (0 eV) without and with Al-doping for the (a) FM and (b) AF solutions. Spin-up/down states are plotted along the positive/negative ordinate.
Co-reporter:Yongmao Cai, Yingjin Wei, Xing Ming, Fei Du, Xing Meng, Chunzhong Wang, Gang Chen
Solid State Communications (May 2011) Volume 151(Issue 10) pp:798-801
Publication Date(Web):1 May 2011
DOI:10.1016/j.ssc.2011.02.031
The phase transition in the perovskite (Pv) SrRuO3 under pressure has been studied by using first-principles calculations based on density functional theory. The post-perovskite (Ppv) phase transition of SrRuO3 will take place under hydrostatic pressure of about 40 GPa. The stability of Ppv- SrRuO3 is justified by the enthalpy calculations, and this phase transition accompanies volume disconnection and magnetic moment collapses. The crystallographic data and the electronic structure of Ppv- SrRuO3 are also predicted. The crystal structure of Ppv- SrRuO3 is similar to that of Ppv- CaRuO3. A non-magnetic ground state is found in Ppv- SrRuO3. The strong hybridization of Ru and O is evident in the electronic structures of both phases. We expect that these results will help further understanding of SrRuO3 under high pressure.Highlights► The post-perovskite (Ppv) phase transition of SrRuO3 has been predicted. ► The Ppv phase transition accompanies volume disconnection and magnetic moment collapses. ► The crystallographic data and the electronic structure of Ppv- SrRuO3 have been revealed.












![zinc (3S,4S,21R)-9-ethenyl-14-ethyl-21-(methoxycarbonyl)-4,8,13,18-tetramethyl-20-oxo-3-(3-oxo-3-{[(2E,7R,11R)-3,7,11,15-tetramethylhexadec-2-en-1-yl]oxy}propyl)-23,25-didehydrophorbine-23,25-diide](http://img.cochemist.com/ccimg/15800/15739-11-4.png)
![zinc (3S,4S,21R)-9-ethenyl-14-ethyl-21-(methoxycarbonyl)-4,8,13,18-tetramethyl-20-oxo-3-(3-oxo-3-{[(2E,7R,11R)-3,7,11,15-tetramethylhexadec-2-en-1-yl]oxy}propyl)-23,25-didehydrophorbine-23,25-diide](http://img.cochemist.com/ccimg/15800/15739-11-4_b.png)