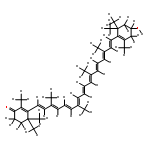
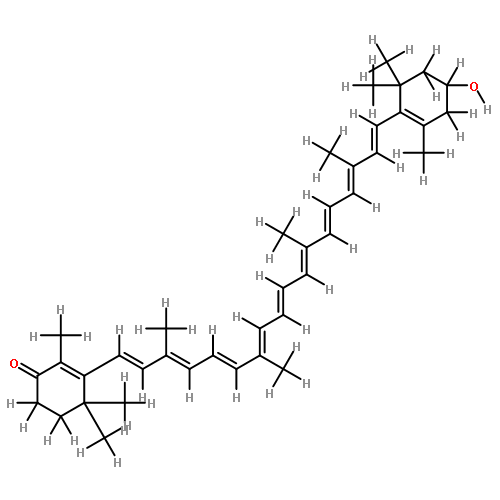
Co-reporter:Yukie Mori
Chemical Physics Letters 2016 Volume 652() pp:184-189
Publication Date(Web):16 May 2016
DOI:10.1016/j.cplett.2016.04.062
•The S0–S2 excitation energy of a carotenoid was calculated with TD-DFT method.•Electrostatic interaction with the protein decreases the excitation energy.•Red shift is caused by the changes of conformation and environmental effect.The orange carotenoid protein, which contains 3′-hydroxyechinenone (hECN), changes color from orange to red when irradiated with blue-green light. In this study, the origins of the color change have been investigated. The conformation of hECN in the red form is more planar than that in the orange form; consequently, the absorption band is red-shifted on conversion from the orange form to the red form. Another source of the red shift is that the electrostatic field generated by the protein in the red form stabilizes the excited state better than that generated by the protein in the orange form.
Co-reporter:Yukie Mori
Journal of Physical Organic Chemistry 2014 Volume 27( Issue 10) pp:803-810
Publication Date(Web):
DOI:10.1002/poc.3339
1,3-Dinitrobenzene radical anion (DNB−), which is a typical mixed valence compound, undergoes intramolecular electron transfer (ET) in solution. It is reported that the ET rates exceed 1010 s−1 in polar aprotic solvent such as acetonitrile. Formulation based on a simple one-dimensional model cannot quantitatively account for the observed ET rates, and further study has been desired for better understanding of the solvent effects on the ET. In the present study, molecular dynamics simulations were performed for DNB− in the vacuum and in acetonitrile solution. In the vacuum, ET was induced by the antisymmetric C–N stretching mode on a timescale of ~100 fs, and the charge transferring between the nitro groups was much less than unity. For the acetonitrile solution, short-timescale and long-timescale simulations were performed using a droplet model of solvated DNB− at 298 K. Although the mean C–N distance in the charged nitro group was longer than that in the vacuum, no ET took place in the short (~150 fs) simulations. The solvent coordinate, which was defined as the difference in the solute–solvent interaction energy between the reactant and the product, significantly fluctuated even in short-time simulations. The reorganization energies in acetonitrile were evaluated on the basis of molecular orbital (MO) calculations, and the ratio of the inner sphere and outer sphere parts, λi/λo, was estimated to be ~0.6. The results suggest that the intramolecular mode and fast solvent mode may play an important role in the present ET reaction. Copyright © 2014 John Wiley & Sons, Ltd.
Co-reporter:Yuichi Masuda, Yukie Mori, and Kazumi Sakurai
The Journal of Physical Chemistry A 2013 Volume 117(Issue 41) pp:10576-10587
Publication Date(Web):September 20, 2013
DOI:10.1021/jp4061297
The proton location and proton transfer (PT) dynamics of a hydrogen bond are under the influence of the static and dynamical properties of the solvent and counterions. In the present study, the N–H distances were determined for salts of 1,8-bis(dimethylamino)naphthalene, DMANH+X– (X– = BPh4–, ClO4–, and Cl–), in acetonitrile (AN) solution, and DMANH+Br– in water by observing the 15N spin–lattice relaxation caused by the 15N–1H magnetic dipolar coupling under assumption that the PT time was shorter than the NH reorientation time (∼10–11 s). The obtained N–H distances decreased in the following order: DMANH+BPh4– > DMANH+ClO4– > DMANH+Br–/H2O > DMANH+Cl–, indicating that interactions with the environment affect the PT potentials. To understand the results at the molecular level, Car–Parrinello molecular dynamics simulations were performed for DMANH+, DMANH+ in water, and DMANH+–Cl– ion-pair in AN. The results of simulation suggest that (1) the N–H distance decreases in the presence of a solvent and counterion; (2) the PT time is probably ∼10–12 s, which confirms the above assumption used for the NMR relaxation data analyses; and (3) fluctuation of the interactions with the solvent or counterion has a significant role in PT. Quantum nuclear effects on the hydrogen bond were also examined.
Co-reporter:Yukie Mori and Keiko Takano
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 31) pp:11090-11098
Publication Date(Web):07 Jun 2012
DOI:10.1039/C2CP41425B
Two-dimensional potential energy surfaces (PESs) were calculated for the degenerate intramolecular proton transfer (PT) in two N–H⋯N hydrogen-bonded systems, (Z)-2-(2-pyridylmethylidene)-1,2-dihydropyridine (1) and monoprotonated di(2-pyridyl) ether (2), at the MP2/cc-pVDZ level of theory. The calculated PES had two minima in both cases. The energy barrier in 1 was higher than the zero-point energy (ZPE) level, while that in 2 was close to the ZPE. Vibrational wavefunctions were obtained by solving time-independent Schrödinger equations with the calculated PESs. The maximum points of the probability density were shifted from the energy minima towards the region where the covalent N–H bond was elongated and the N⋯N distance shortened. The effects of a polar solvent on the PES were investigated with the continuum or cluster models in such a way that the solute–solvent electrostatic interactions could be taken into account under non-equilibrated conditions. A solvated contact ion-pair was modelled by a cluster consisting of one cation 2, one chloride ion and 26 molecules of acetonitrile. The calculation with this model suggested that the bridging proton is localised in the deeper well due to the significant asymmetry of the PES and the high potential barrier.
Co-reporter:Yukie Mori, Keiko Takano
Chemical Physics Letters 2011 Volume 511(4–6) pp:251-255
Publication Date(Web):5 August 2011
DOI:10.1016/j.cplett.2011.06.030
Abstract
The reaction mechanisms for cycloreversion of anthracene–benzene and naphthalene–benzene [4 + 4] cycloadducts have been studied by CASPT2//CASSCF calculations. Upon UV excitation, the initially populated 1(π, π∗) state is converted to the 1(σ, σ∗) state, which undergoes the cycloreversion adiabatically to give the product in the singlet excited state. The barrierless and downhill nature of the potential energy curve accounts for the high quantum yield of anthracene in the 1La state. In the naphthalene–benzene system, the 1Lb state of naphthalene is formed via internal conversion from the 1La state that is directly generated in the reaction.
Co-reporter:Yukie Mori, Keiko Takano
Journal of Photochemistry and Photobiology A: Chemistry 2011 Volume 219(2–3) pp:278-284
Publication Date(Web):15 April 2011
DOI:10.1016/j.jphotochem.2011.03.004
The potential energy surfaces (PESs) were investigated for di-π-methane rearrangement of 4-phenyl-4H-pyran at CASSCF(12,12)/6-31G(d) and multireference second-order perturbation theory. The three-step mechanism proposed by Zimmerman was confirmed for the reaction from the excited triplet state. The minimum energy path indicated that the initial σ-bond formation takes place from the 3(π,π*) state localized at the C2C3 olefinic moiety to give a primary phenyl-bridged biradical (3BR1). This biradical is immediately converted to a 1,3-biradical (3BR2). The intersystem crossing to 1BR2 can take place in the vicinity of the minimum energy crossing point due to the spin-orbit coupling interaction. 1BR2 is rather different from 3BR2 in geometrical and electronic structures and is regarded as a biradicaloid with zwitterionic character. The subsequent ring-closure of 1BR2 is almost barrierless and yields the expected product, 6-endo-phenyl-2-oxabicyclo[3.1.0]hexene. These results provide deeper insight into the previous experimental observations regarding the photochemical reactions of related compounds.Highlights► The PESs were studied for di-π-methane rearrangement. ► The reaction proceeds via triplet biradical. ► After ISC, formation of the product is barrierless. ► The results agree with the experiments.
Co-reporter:Yukie Mori and Keiko Takano
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 31) pp:NaN11098-11098
Publication Date(Web):2012/06/07
DOI:10.1039/C2CP41425B
Two-dimensional potential energy surfaces (PESs) were calculated for the degenerate intramolecular proton transfer (PT) in two N–H⋯N hydrogen-bonded systems, (Z)-2-(2-pyridylmethylidene)-1,2-dihydropyridine (1) and monoprotonated di(2-pyridyl) ether (2), at the MP2/cc-pVDZ level of theory. The calculated PES had two minima in both cases. The energy barrier in 1 was higher than the zero-point energy (ZPE) level, while that in 2 was close to the ZPE. Vibrational wavefunctions were obtained by solving time-independent Schrödinger equations with the calculated PESs. The maximum points of the probability density were shifted from the energy minima towards the region where the covalent N–H bond was elongated and the N⋯N distance shortened. The effects of a polar solvent on the PES were investigated with the continuum or cluster models in such a way that the solute–solvent electrostatic interactions could be taken into account under non-equilibrated conditions. A solvated contact ion-pair was modelled by a cluster consisting of one cation 2, one chloride ion and 26 molecules of acetonitrile. The calculation with this model suggested that the bridging proton is localised in the deeper well due to the significant asymmetry of the PES and the high potential barrier.