.jpg)
Amphiphilic hyperbranched copolymer chains made of large hyperbranched poly(acrylic acid) cores grafted with short polystyrene stickers (HB-PAAn-g-PSn + 1) with different n values (n = 1, 10, 47) were well prepared and confirmed by size exclusion chromatography, Fourier transform infrared spectroscopy and 1H nuclear magnetic resonance. The study on the interchain association behavior of these amphiphilic chains indicates that larger HB-(PAA)n-g-(PS)n + 1 copolymer chains have a less tendency to undergo interchain association. Moreover, the simple vial-inversion and rheological experiments show that the apparent critical gel concentration (Cg) decreases with n, but no sol–gel transition was observed for triblock PS-PAA-PS even when the concentration is up to 200 g L−1. Further transmission electron microscopy study of the latex particles prepared with HB-(PAA)n-g-(PS)n + 1 as surfactant reveals that the latex particles are spherical and narrowly dispersed; while the measured latex particle number (Np) indicates the surfactant efficiency of HB-(PAA)47-g-(PS)48 is poorer than that of triblock PS-PAA-PS (n = 1). Finally, pyrene solubilization measurement shows the solubilization efficiency of HB-(PAA)n-g-(PS)n + 1 copolymers decreases with n, consistent with the previous observed interchain association result. The present study demonstrates that both the chain topology and the styrene weight fraction dominates the final solution properties of amphiphilic HB-(PAA)n-g-(PS)n + 1 chains in aqueous solution. © 2013 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2014, 52, 128–138
Long-subchain hyperbranched polystyrene (lsc-hp PSt) with uniform subchain length was obtained through copper-catalyzed azide-alkyne cycloaddition click chemistry from seesaw macromonomer of PSt having one alkynyl group anchored at the chain centre and two azido group attached to both chain ends [alkynyl-(PSt-N3)2]. After precipitation fraction, different portions of lsc-hp PSt having narrow overall molecular weight distribution were obtained for further grafting with alkynyl-capped poly(N-isopropylacrylamide) (alkynyl-PNIPAM), which was obtained via single-electron transfer living radical polymerization of NIPAM with propargyl 2-bromoisobutyrate as the initiator and grafted onto the peripheral azido groups of lsc-hp PSt via click chemistry. Thus, amphiphilic lsc-hp PSt grafted with PNIPAM chains (lsc-hp PSt-g-PNIPAM) was obtained and would have star-like conformation in tetrahydrofuran (THF). By replacing THF with water, lsc-hp PSt-g-PNIPAM was dissolved at molecular level in aqueous solution due to the hydrophilicity of PNIPAM and exhibited thermal induced shrinkage of PNIPAM arms. The water-insoluble lsc-hp PSt would collapse densely and could be served as a reservoir to absorb hydrophobic chemicals in aqueous solution. The influence of overall molecular weight of lsc-hp PSt on the absorption of pyrene was studied. © 2013 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2013
Hollow mesoporous silica nanoparticles (HMSNs) grafted with a photo-responsive copolymer containing coumarin groups were successfully prepared. With uniform polystyrene nanoparticles and cetyltrimethylammonium bromide correspondingly as the template of core and channel, HMSNs were made from tetraethyloxysilane in alkalic condition. Epoxy groups were introduced onto the outer surface of HMSNs with γ-(2,3-epoxypropoxy)propyltrimethoxysilane and converted into azido groups with sodium azide, resulting in azido-functionalized HMSNs (azido-HMSNs). Meanwhile, single-electron transfer-living radical copolymerization of methyl methacrylate (MMA) and 7-(2-methacryloyloxy)-4-methylcoumarin (CMA) with propargyl 2-bromoisobutyrate as the initiator produced alkynyl-capped P(MMA-co-CMA) [alkynyl-P(MMA-co-CMA)]. Finally, photo-responsive HMSNs grafted with P(MMA-co-CMA) [HMSN-g-P(MMA-co-CMA)] was achieved through the click reaction between azido-HMSNs and alkynyl-P(MMA-co-CMA). Different techniques such as transmission electron microscopy, Fourier transform infrared spectroscopy, and thermal gravimetric analysis confirmed the successful preparation of the resultant hybrid nanoparticles and their intermediates. Because of its hollow core, mesoporous shell channels and light responsiveness, the coumarin-modified HMSNs would be an interesting nano-vehicle for guest molecules. Thus, the loading and release of pyrene with HMSN-g-P(MMA-co-CMA) was studied. © 2013 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 3791–3799
Through atom transfer radical polymerization of styrene with 1,3-dibromomethyl-5-propargyloxy-benzene as initiator followed by the conversion of bromine end-groups into azide end-groups, well-defined seesaw-type polystyrene (PSt) macromonomers with two molecular weights (Mn = 8.0 and 28.0 k) were obtained. Thus, a series of long-subchain hyperbranched (lsc-hp) PSt with high overall molar masses and regular subchain lengths were obtained via copper-catalyzed azide–alkyne cycloaddition click chemistry performed in THF and DMF, respectively. The polycondensation of seesaw-type macromonomers was monitored by gel permeation chromatography. Because DMF is the reaction medium with higher polarity, click reaction proceeds more easily in DMF. Therefore, the growth of lsc-hp PSt in DMF has faster rate than that in THF for the shorter seesaw-type macromonomer (Seesaw-8k). However, THF is the solvent with better solubility to PSt and leads to looser conformation of PSt chains. Thus, for the longer seesaw macromonomer (Seesaw-28k), lsc-hp PSt in THF has higher overall molar mass. As well, the self-cyclization of seesaw-type macromonomers also depends on both solvent and molar mass of macromonomer. The self-cyclization degrees of Seesaw-8k in DMF and THF are almost the same while that of Seesaw-28k macromonomer is obviously lower in THF. The experimental results suggest a physical consideration to control the growth of hyperbranched polymers. © 2012 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Through reversible addition-fragmentation chain transfer (RAFT) polymerization of t-butyl acrylate (tBA) and RAFT copolymerization of 2-dimethylaminoethyl methacrylate (DMAEMA) with poly(ethylene glycol) methyl ether methacrylate (PEGMEMA), block-comb copolymer of PtBA-b-P(PEGMEMA-co-DMAEMA) was prepared. After the self-assembly of PtBA-b-P(PEGMEMA-co-DMAEMA) into core-shell spherical micelles, P(PEGMEMA-co-DMAEMA) segments of the shell was crosslinked with 1,2-bis(2-iodoethoxy)ethane and the core of PtBA was selectively hydrolysized with trifluoroacetic acid. Thus, zwitterionic shell-crosslinked micelles with positively charged outer shell and negatively charged inner core were obtained. Dynamic light scattering, transmission electron microscope, Zeta potential measurement, and nuclear magnetic resonance were used to confirm the formation of the zwitterionic shell-crosslinked micelles. They showed the excellent resistance to the variation of pH value and possessed the positive values throughout the whole range of pH range even if the carboxylic groups of the micelles was much more than ammonium groups. © 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2011
Dual thermo- and pH-sensitive network-grafted hydrogels made of poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) network and poly(N-isopropylacrylamide) (PNIPAM) grafting chains were successfully synthesized by the combination of atom transfer radical polymerization (ATRP), reversible addition-fragmentation chain transfer (RAFT) polymerization, and click chemistry. PNIPAM having two azide groups at one chain end [PNIPAM-(N3)2] was prepared with an azide-capped ATRP initiator of N,N-di(β-azidoethyl) 2-chloropropionylamide. Alkyne-pending poly(N,N-dimethylaminoethyl methacrylate-co-propargyl acrylate) [P(DMAEMA-co-ProA)] was obtained through RAFT copolymerization using dibenzyltrithiocarbonate as chain transfer agent. The subsequent click reaction led to the formation of the network-grafted hydrogels. The influences of the chemical composition of P(DMAEMA-co-ProA) on the properties of the hydrogels were investigated in terms of morphology and swelling/deswelling kinetics. The dual stimulus-sensitive hydrogels exhibited fast response, high swelling ratio, and reproducible swelling/deswelling cycles under different temperatures and pH values. The uptake and release of ceftriaxone sodium by these hydrogels showed both thermal and pH dependence, suggesting the feasibility of these hydrogels as thermo- and pH-dependent drug release devices. © 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2011
Reducibly degradable hydrogels of poly(N-isopropylacrylamide) (PNIPAM) and poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) were synthesized by the combination of reversible addition-fragmentation chain transfer (RAFT) polymerization and click chemistry. The alkyne-pending copolymer of PNIPAM or PDMAEMA was obtained through RAFT copolymerization of propargyl acrylate with NIPAM or DMAEMA. Bis-2-azidyl-isobutyrylamide of cystamine (AIBCy) was used as the crosslinking reagent to prepare reducibly degradable hydrogels by click chemistry. The hydrogels exhibited temperature or pH stimulus-responsive behavior in water, with rapid response, high swelling ratio, and reproducible swelling/shrinkage cycles. The loading and release of ceftriaxone sodium proved the feasibility of the hydrogels as the stimulus-responsive drug delivery system. Furthermore, the presence of disulfide linkage in AIBCy favored the degradation of hydrogels in the reductive environment. © 2010 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 48: 3604–3612, 2010
Well-defined H-shaped pentablock copolymers composed of poly(N-isopropylacrylamide) (PNIPAM), poly(N,N-dimethylaminoethylacrylamide) (PDMAEMA), and poly(ethylene glycol) (PEG) with the chain architecture of (A/B)-b-C-b-(A/B) were synthesized by the combination of single-electron-transfer living radical polymerization, atom-transfer radical polymerization, and click chemistry. Single-electron-transfer living radical polymerization of NIPAM using α,ω azide-capped PEG macroinitiator resulted in PNIPAM-b-PEG-b-PNIPAM with azide groups at the block joints. Atom-transfer radical polymerization of DMAEMA initiated by propargyl 2-chloropropionate gave out α-capped alkyne-PDMAEMA. The H-shaped copolymers were finally obtained by the click reaction between PNIPAM-b-PEG-b-PNIPAM and alkyne-PDMAEMA. These copolymers were used to prepare stable colloidal gold nanoparticles (GNPs) in aqueous solution without any external reducing agent. The formation of GNPs was affected by the length of PDMAEMA block, the feed ratio of the copolymer to HAuCl4, and the pH value. The surface plasmon absorbance of these obtained GNPs also exhibited pH and thermal dependence because of the existence of PNIAPM and PDAMEMA blocks. © 2010 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2010
A hetero-arm star polymer, polystyrene-poly(N-isopropylacrylamide)- poly(2-(dimethylamino)ethylmethacrylate) (PSt-PNIPAM-PDMAEMA), was synthesized by “clicking” the alkyne group at the junction of PSt-b-PNIPAM diblock copolymer onto the azide end-group of PDMAEMA homopolymer via 1,3-dipolar cycloaddition. The resultant polymer was characterized by gel permeation chromatography, proton nuclear magnetic resonance spectroscopy and Fourier transform infrared spectroscopy. PSt-PNIPAM-PDMAEMA micelles with PSt block as core and PNIPAM and PDMAEMA blocks as shell were formed when adding the copolymer solution in THF into 10 folds of water. Lower critical solution temperature (LCST) of PNIPAM and PDMAEMA homopolymer is 32 °C for PNIPAM and 40 to 50 °C for PDMAEMA, respectively. Upon continuous heating through their LCSTs, PSt-PNIPAM-PDMAEMA core-shell micelles exhibited two-stage thermally induced collapse. The first-stage collapse, from 20 to 34 °C, is ascribed to the shrinkage of PNIPAM chains; and the second-stage collapse, from 38 to 50 °C, is due to the shrinkage of PDMAEMA chains. Dynamic light scattering was used to confirm the double phase transitions. © 2008 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 786–796, 2009
A hetero-arm star polymer, poly(ethylene glycol)-poly(N-isopropylacrylamide)-poly(L-lysine) (PEG-PNIPAM-PLys), was synthesized by “clicking” the azide group at the junction of PEG-b-PNIPAM diblock copolymer with the alkyne end-group of poly(L-lysine) (PLys) homopolymer via 1,3-dipolar cycloaddition. The resultant polymer was characterized by gel permeation chromatography, proton nuclear magnetic resonance, and Fourier transform infrared spectroscopes. Surprisingly, the PNIPAM arm of this hetero-arm star polymer nearly lose its thermal responsibility. It is found that stable polyelectrolyte complex micelles are formed when mixing the synthesized polymer with poly(acrylic acid) (PAA) in water. The resultant polyelectrolyte complex micelles are core-shell spheres with the ion-bonded PLys/PAA chains as core and the PEG and PNIPAM chains as shell. The PNIPAM shell is, as expected, thermally responsive. However, its lower critical solution temperature is shifted to 37.5 °C, presumably because of the existence of hydrophilic components in the micelles. Such star-like PEG-PNIPAM-PLys polymer with different functional arms as well as its complexation with anionic polymers provides an excellent and well-defined model for the design of nonviral vectors to deliver DNA, RNA, and anionic molecular medicines. © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 1450–1462, 2009
Degradable hydrogels crosslinked with disulfide bonds were prepared by Michael addition between amine groups of branched polyethylenimine and carbon–carbon double bonds of N,N′-bis(acryloyl)cystamine. The influences of the chemical composition of the resulted hydrogels on their properties were examined in terms of morphology, surface area, swelling kinetics, and degradation. The hydrogels were uniformly crosslinked and degraded into water-soluble polymers in the presence of the reducing agent of dithiothreitol, which improved the control over the release of encapsulated drug. The degradation of hydrogels can trigger the release of encapsulated molecules, as well as facilitate the removal of empty vehicles. Results obtained from in vitro drug release suggested that the disulfide crosslinked hydrogels exhibited an accelerated release of encapsulated drug in dithiothreitol-containing PBS buffer solution. Moreover, the drug release rate decreased gradually with increasing crosslinking density. © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 4074–4082, 2009
Aqueous reversible addition-fragmentation chain transfer (RAFT) cryopolymerizations of N,N-dimethylacrylamide (DMA) and N-isopropylacrylamide (NIPAM) with potassium persulfate/sodium ascorbate as redox initiators were performed at −15 °C. For the homopolymerizations, water-soluble chain transfer agents (CTAs) of 2-(1-carboxy-1-methylethyl-sulfanylthiocarbonylsulfanyl)-2-methylpropionic acid and 2-dodecylsulfanylthiocarbonylsulfanyl-2-methylpropionyl-capped methoxy poly(ethylene glycol) were used. For the sequential block copolymerizations, the obtained trithiocarbonate-functionalized polymers were used as macro-CTAs. Although well-defined homo and block polymers of DMA and NIPAM were synthesized and these RAFT cryopolymerizations were well controlled, their behavior depended on the monomers and CTAs. The polymerization kinetic and polymer structure were studied by proton nuclear magnetic resonance analysis and gel permeation chromatography measurement. Poly(N,N-dimethylacrylamide)-based cryogels crosslinked with reductively cleavable disulfide-containing diacrylamide, N,N′-bisacryloylcystamine, were synthesized via RAFT cryopolymerization. Scanning electron microscopy observation revealed that the porous structure of cryogels depended on the CTA used. © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2009
Twin-tail tadpole-shaped hydrophillic copolymers composed of cyclic poly(ethylene gycol) (PEG) and two linear poly(N-isopropylacrylamide) (PNIPAM) chains have been successfully synthesized by the combination of single-electron-transfer living radical polymerization and click chemistry under high concentration. Click cycloaddition reaction occurred between linear PNIPAM-b-PEG-b-PNIPAM with two azide groups at block junctions and dipropargyl oxalylate with high yield and efficiency. The resulting intermediates and the targeted polymers were characterized by proton nuclear magnetic resonance, fourier transform infrared spectroscopy, and gel permeation chromatography. The thermal phase transition behaviors of twin-tail tadpole-shaped polymers and their linear precursors were investigated by temperature-dependent turbidity measurements, micro differential scanning calorimetry, and laser light scattering. The twin-tail tadpole-shaped polymers possess higher critical solution temperature (LCST) and smaller average aggregate size compared with their linear precursors with the same molecular weight. The above differences in the thermal phase transition behaviors should be due to the repulsive forces caused by the ring topology, which prohibited the intermolecular association. © 2009 Wiley Periodicals, © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2009
Pyrrolyl-capped poly(N-isopropylacrylamide) macromonomers (Py-PNIPAM) were prepared through reversible addition-fragmentation-transfer polymerization with benzyl 1-pyrrolylcarbodithioate as chain-transfer agent. Polymerizations of Py-PNIPAM with/without pyrrole using AgNO3 as oxidizing agent and dimethylforamide as solvent resulted in graft copolymers of polypyrrole-graft-poly(N-isopropylacrylamide) (PPy-g-PNIPAM) as well as silver nanoparticles, leading to the formation of PPy-g-PNIPAM/silver nanocomposites. The resulting nanocomposites were soluble in water when the content of PPy was low, and when the molar ratio of Py/Py-PNIPAM increased to 30, the resulting products became insoluble in water. The resulting nanocomposites had special optical properties because of PPy as well as the temperature-responsible PNIPAM. The chemical structure and composition of nanocomposite were characterized by 1H nuclear magnetic resonance spectroscopy, gel permeation chromatograms, fourier transform infrared spectroscopy, and X-ray diffraction. Their optical properties were characterized by UV–vis and fluorescence spectroscopy. © 2008 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 46: 6950–6960, 2008
Poly(styrene-b-N-isopropylacrylamide) (PSt-b-PNIPAM) with dithiobenzoate terminal group was synthesized by reversible addition-fragmentation-transfer polymerization. The dithiobenzoate terminal group was converted into thiol terminal group with NaBH4, resulting thiol-terminated PSt-b-PNIPAM-SH. After PSt-b-PNIPAM-SH assembled into core-shell micelles in aqueous solution, gold nanoparticles were in situ surface-linked onto the micelles through the reduction of gold precursor anions with NaBH4. Thus, temperature responsive core/shell micelles of PSt-b-PNIPAM surface-linked with gold nanoparticles (PSt-b-PNIPAM-Au micelles) were obtained. Transmission Electron Microscopy revealed the successful linkage of gold nanoparticles and the dependence of the number of gold nanoparticles per micelle on the molar ratio of HAuCl4 to thiol group of PSt-b-PNIPAM. Dynamic Light Scattering analysis demonstrated thermo-responsive behavior of PSt-b-PNIPAM-Au micelles. Changing the temperature of PSt-b-PNIPAM-Au micelles led to the shrinkage of PNIPAM shell and allowed to tune the distance between gold nanoparticles. Ultraviolet–visible (UV–vis) spectroscopy clearly showed the reversible modulation of UV–vis absorbance of PSt-b-PNIPAM-Au micelles upon heating and cooling. © 2007 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 45: 5156–5163, 2007
Summary: Copolymerizations of St and NIPAM have been carried out through interfacial-initiated microemulsion polymerization in a frozen state. FT-IR and NMR spectroscopies confirm the occurrence of copolymerization between the two monomers. DSC analysis shows the existence of two glass transition temperatures of the resultant copolymers. The micellization of the copolymers is investigated by DLS and the temperature-responsive behavior of the resultant micelles is observed. DSC and DLS results reveal the block feature of the obtained copolymers. Thus amphiphilic poly(styrene-block-N-isopropylacrylamide) is prepared by a one-step interfacial-initiated microemulsion polymerization.
Interfacially initiated microemulsion copolymerizations of n-butyl methacrylate (BMA) and N-vinyl pyrrolidone (NVP) by the redox initiation couple of benzoyl peroxide and ferrous sulfate were carried out with Tween 80 and n-butanol as the surfactant and cosurfactant, respectively. Fourier transform infrared spectroscopy and X-ray photoelectron spectroscopy were recorded to analyze the chemical composition of the latex particles. Transmission electron microscopy was used to observe the particle morphology and dynamic light scattering to determine the particle size. The results demonstrated that interfacially initiated microemulsion polymerization prompted the copolymerization of the water-soluble NVP monomer with the oil-soluble BMA monomer to form core–shell nanoparticles. The influence of the surfactant concentration, BMA amount, and temperature on the particle size and polymerization rate was investigated. © 2006 Wiley Periodicals, Inc. J Appl Polym Sci 101: 3751–3757, 2006
Composite polymer particles with hydrophobic polystyrene (PSt) as the core and hydrophilic poly(methacrylic acid) (PMAA) as the shell were prepared through two-stage emulsion polymerization without any surfactant. In the first step, narrowly distributed PSt seed particles were prepared by surfactant-free emulsion polymerization with 2,2′-azobis(2-methylpropionamide) dihydrochloride (AMPA) as the initiator. In the second step, hydrophilic PMAA shells were fabricated onto PSt seed particles through redox interfacial-initiated seeded emulsion polymerization with cumyl hydroperoxide (CHPO)/ferrous sulfate/ethylenediaminetetraacetic acid (EDTA)/sodium formaldehydesulfoxylate (SFS), where the initiation took place mainly at the interface between PSt seed particles and the aqueous medium. The composite particles were characterized with transmission electron microscopy, fourier transform infrared spectroscopy and dynamic light scattering, and the results show that a core/shell structure was successfully built. Hydrodynamic radius (Rh) of the composite particles increased with the amount of polymerized monomers in the seeded emulsion polymerization. Copyright © 2006 Society of Chemical Industry
Narrowly distributed nanoparticles of poly (n-butyl methacrylate-co-vinyl pyrrolidone) were prepared through microemulsion polymerization with a nonionic surfactant of Tween-80 as emulsifier (6 wt % of the latex) and n-butanol as coemulsifier. The polymerizations were initiated with benzoylperoxide (BPO), potassium persulfate (KPS), KPS/ferric sulfate (FeSO4), and BPO/FeSO4, respectively, where the initiation in the case of BPO/FeSO4 took place mainly at the interphase between the oil phase and the reaction media. Namely, this interfacial-initiated microemulsion polymerization resulted in larger particles with relatively narrower particle size distribution as well as higher limiting monomer conversion but lower polymerization rate compared with the polymerization initiated with KPS/FeSO4. In this article, the influences of initiation method, monomer ratio, and addition of water-soluble components on microemulsion polymerization and latex particle size were studied to discuss the mechanism of interfacial-initiated microemulsion polymerization. © 2004 Wiley Periodicals, Inc. J Appl Polym Sci 92: 2334–2340, 2004
Seeded emulsion polymerization of methyl methacrylate (MMA) or styrene (ST) was carried in the presence of different vinyl-containing polysiloxane latices (SV-*) and the core-shell particles with poly(methyl methacrylate) (PMMA) or polystyrene (PST), as the shells were obtained under different polymerization conditions. Besides the compatibility of the vinyl monomer and its polymer with polysiloxane and the reaction between vinyl monomer with vinyl group of polysiloxane, the content of vinyl group of seed polysiloxane has influence on the morphology and component of the resulted composite particles. The mechanism for the formation of core-shell structure is discussed. © 2001 John Wiley & Sons, Inc. J Appl Polym Sci 80: 2752–2758, 2001
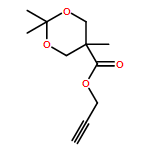
![Propanoic acid, 3-(2-bromo-2-methyl-1-oxopropoxy)-2-[(2-bromo-2-methyl-1-oxopropoxy)methyl]-2-methyl-, 2-propyn-1-yl ester](/data/chemimg/117800/949023-35-2.png)
![Propanoic acid, 3-(2-bromo-2-methyl-1-oxopropoxy)-2-[(2-bromo-2-methyl-1-oxopropoxy)methyl]-2-methyl-, 2-propyn-1-yl ester](/data/chemimg/117800/949023-35-2_b.png)