Co-reporter:Benjamin A. Gilston;Eric P. Skaar
Science China Life Sciences 2016 Volume 59( Issue 8) pp:792-801
Publication Date(Web):2016 August
DOI:10.1007/s11427-016-5088-4
The S100 proteins are a unique class of EF-hand Ca2+ binding proteins distributed in a cell-specific, tissue-specific, and cell cycle-specific manner in humans and other vertebrates. These proteins are distinguished by their distinctive homodimeric structure, both intracellular and extracellular functions, and the ability to bind transition metals at the dimer interface. Here we summarize current knowledge of S100 protein binding of Zn2+, Cu2+ and Mn2+ ions, focusing on binding affinities, conformational changes that arise from metal binding, and the roles of transition metal binding in S100 protein function.
Co-reporter:Norie Sugitani ; Steven M. Shell ; Sarah E. Soss
Journal of the American Chemical Society 2014 Volume 136(Issue 31) pp:10830-10833
Publication Date(Web):July 21, 2014
DOI:10.1021/ja503020f
Xeroderma pigmentosum complementation group A (XPA) protein plays a critical role in the repair of DNA damage via the nucleotide excision repair (NER) pathway. XPA serves as a scaffold for NER, interacting with several other NER proteins as well as the DNA substrate. The critical importance of XPA is underscored by its association with the most severe clinical phenotypes of the genetic disorder Xeroderma pigmentosum. Many of these disease-associated mutations map to the XPA98–219 DNA-binding domain (DBD) first reported ∼20 years ago. Although multiple solution NMR structures of XPA98–219 have been determined, the molecular basis for the interaction of this domain with DNA is only poorly characterized. In this report, we demonstrate using a fluorescence anisotropy DNA-binding assay that the previously reported XPA DBD binds DNA with substantially weaker affinity than the full-length protein. In-depth analysis of the XPA sequence suggested that the original DBD construct lacks critical basic charge and helical elements at its C-terminus. Generation and analysis of a series of C-terminal extensions beyond residue 219 yielded a stable, soluble human XPA98–239 construct that binds to a Y-shaped ssDNA–dsDNA junction and other substrates with the same affinity as the full-length protein. Two-dimensional 15N–1H NMR suggested XPA98–239 contains the same globular core as XPA98–219 and likely undergoes a conformational change upon binding DNA. Together, our results demonstrate that the XPA DBD should be redefined and that XPA98–239 is a suitable model to examine the DNA binding activity of human XPA.
Co-reporter:Michael D. Feldkamp, Aaron C. Mason, Brandt F. Eichman, and Walter J. Chazin
Biochemistry 2014 Volume 53(Issue 18) pp:
Publication Date(Web):April 15, 2014
DOI:10.1021/bi500252w
SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A-like1 (SMARCAL1) is a recently identified DNA damage response protein involved in remodeling stalled replication forks. The eukaryotic single-strand DNA binding protein replication protein A (RPA) recruits SMARCAL1 to stalled forks in vivo and facilitates regression of forks containing leading strand gaps. Both activities are mediated by a direct interaction between an RPA binding motif (RBM) at the N-terminus of SMARCAL1 and the C-terminal winged-helix domain of the RPA 32 kDa subunit (RPA32C). Here we report a biophysical and structural characterization of the SMARCAL1–RPA interaction. Isothermal titration calorimetry and circular dichroism spectroscopy revealed that RPA32C binds SMARCAL1-RBM with a Kd of 2.5 μM and induces a disorder-to-helix transition. The crystal structure of RPA32C was refined to 1.4 Å resolution, and the SMARCAL1-RBM binding site was mapped on the structure on the basis of nuclear magnetic resonance chemical shift perturbations. Conservation of the interaction surface to other RBM-containing proteins allowed construction of a model for the RPA32C/SMARCAL1-RBM complex. The implications of our results are discussed with respect to the recruitment of SMARCAL1 and other DNA damage response and repair proteins to stalled replication forks.
Co-reporter:Sarah E. Soss, Rachel E. Klevit, and Walter J. Chazin
Biochemistry 2013 Volume 52(Issue 17) pp:
Publication Date(Web):April 3, 2013
DOI:10.1021/bi3015949
Post-translational modification of proteins with ubiquitin is mediated by dynamic multienzyme machinery (E1, E2, and E3). E3 ubiquitin ligases play a key role acting as both scaffolds to bring reactants together and activators to catalyze ubiquitin (Ub) transfer from E2∼Ub conjugates to substrates. Our recent studies provided insights into the mechanism of the activation event; binding of an E3 to an E2∼Ub conjugate was found to affect the motions of E2∼Ub and allosterically stimulate Ub transfer. This proposed mechanism implies that the dynamics of the conjugate, which has been shown to occupy a wide range of E2∼Ub orientations, will be altered significantly upon binding of E3. To directly assess the effect of E3 binding on E2∼Ub dynamics, we undertook an in-depth comparative analysis of 15N nuclear magnetic resonance relaxation of UbcH5c∼Ub in the absence and presence of the E3 ligase, E4B. Challenges encountered in deciphering interdomain motions for this ternary complex are discussed along with the limitations of the current approaches. Notably, although a reduction in interdomain dynamics of UbcH5c∼Ub is observed upon binding to E4B, Ub retains an extensive degree of flexibility. These results provide strong support for our dynamic model of a significant orientational bias of Ub toward a more closed conformation in the E3/E2∼Ub complex.
Co-reporter:Michael D. Feldkamp, Andreas O. Frank, J. Phillip Kennedy, James D. Patrone, Bhavatarini Vangamudi, Alex G. Waterson, Stephen W. Fesik, and Walter J. Chazin
Biochemistry 2013 Volume 52(Issue 37) pp:
Publication Date(Web):August 20, 2013
DOI:10.1021/bi400542z
Replication protein A (RPA) is the primary single-stranded DNA (ssDNA) binding protein in eukaryotes. The N-terminal domain of the RPA70 subunit (RPA70N) interacts via a basic cleft with a wide range of DNA processing proteins, including several that regulate DNA damage response and repair. Small molecule inhibitors that disrupt these protein–protein interactions are therefore of interest as chemical probes of these critical DNA processing pathways and as inhibitors to counter the upregulation of DNA damage response and repair associated with treatment of cancer patients with radiation or DNA-damaging agents. Determination of three-dimensional structures of protein–ligand complexes is an important step for elaboration of small molecule inhibitors. However, although crystal structures of free RPA70N and an RPA70N–peptide fusion construct have been reported, RPA70N–inhibitor complexes have been recalcitrant to crystallization. Analysis of the P61 lattice of RPA70N crystals led us to hypothesize that the ligand-binding surface was occluded. Surface reengineering to alter key crystal lattice contacts led to the design of RPA70N E7R, E100R, and E7R/E100R mutants. These mutants crystallized in a P212121 lattice that clearly had significant solvent channels open to the critical basic cleft. Analysis of X-ray crystal structures, target peptide binding affinities, and 15N–1H heteronuclear single-quantum coherence nuclear magnetic resonance spectra showed that the mutations do not result in perturbations of the RPA70N ligand-binding surface. The success of the design was demonstrated by determining the structure of RPA70N E7R soaked with a ligand discovered in a previously reported molecular fragment screen. A fluorescence anisotropy competition binding assay revealed this compound can inhibit the interaction of RPA70N with the peptide binding motif from the DNA damage response protein ATRIP. The implications of the results are discussed in the context of ongoing efforts to design RPA70N inhibitors.
Co-reporter:Steven M. Damo;Wesley J. Murphy;Thomas E. Kehl-Fie;Christine Betz;Laura Hench;Günter Fritz;Marilyn E. Holt;Eric P. Skaar;Yaofang Zhang;Norie Sugitani;Subodh Rathi
PNAS 2013 Volume 110 (Issue 10 ) pp:3841-3846
Publication Date(Web):2013-03-05
DOI:10.1073/pnas.1220341110
The S100A8/S100A9 heterodimer calprotectin (CP) functions in the host response to pathogens through a mechanism termed “nutritional
immunity.” CP binds Mn2+ and Zn2+ with high affinity and starves bacteria of these essential nutrients. Combining biophysical, structural, and microbiological
analysis, we identified the molecular basis of Mn2+ sequestration. The asymmetry of the CP heterodimer creates a single Mn2+-binding site from six histidine residues, which distinguishes CP from all other Mn2+-binding proteins. Analysis of CP mutants with altered metal-binding properties revealed that, despite both Mn2+ and Zn2+ being essential metals, maximal growth inhibition of multiple bacterial pathogens requires Mn2+ sequestration. These data establish the importance of Mn2+ sequestration in defense against infection, explain the broad-spectrum antimicrobial activity of CP relative to other S100
proteins, and clarify the impact of metal depletion on the innate immune response to infection.
Co-reporter:Walter J. Chazin
Accounts of Chemical Research 2011 Volume 44(Issue 3) pp:171
Publication Date(Web):February 11, 2011
DOI:10.1021/ar100110d
The EF hand, a helix−loop−helix structure, is one of the most common motifs found in animal genomes, and EF-hand Ca2+-binding proteins (EFCaBPs) are widely distributed throughout the cell. However, researchers remain confounded by a lack of understanding of how peptide sequences code for specific functions and by uncertainty about the molecular mechanisms that enable EFCaBPs to distinguish among many diverse cellular targets. Such knowledge could define the roles of EFCaBPs in health and disease and ultimately enable control or even design of Ca2+-dependent functions in medicine and biotechnology. In this Account, we describe our structural and biochemical research designed to understand the sequence-to-function relationship in EFCaBPs.The first structural goal was to define conformational changes induced by binding Ca2+, and our group and others established that solution NMR spectroscopy is well suited for this task. We pinpointed residues critical to the differences in Ca2+ response of calbindin D9k and calmodulin (CaM), homologous EFCaBPs from different functional classes, by using direct structure determination with site-directed mutagenesis and protein engineering. Structure combined with biochemistry provided the foundation for identifying the fundamental mechanism of cooperativity in the binding of Ca2+ ions: this cooperativity provides EFCaBPs with the ability to detect the relatively small changes in concentration that constitute Ca2+ signals. Using calbindin D9k as a model system, studies of the structure and fast time scale dynamics of each of the four ion binding states in a typical EF-hand domain provided direct evidence that site−site communication lowers the free energy cost of reorganization for binding the second ion.Our work has also extended models of how EFCaBPs interact with their cellular targets. We determined the unique dimeric architecture of S100 proteins, a specialized subfamily of EFCaBPs found exclusively in vertebrates. We described the implications for how these proteins transduce signals and went on to characterize interactions with peptide fragments of important cellular targets. Studies of the CaM homolog centrin revealed novel characteristics of its binding of Ca2+ and its interaction with its cellular target Kar1. These results provided clear examples of how subtle differences in sequence fine-tune EFCaBPs to interact with their specific targets.The structural approach stands at a critical crossroad, shifting in emphasis from descriptive structural biochemistry to integrated biology and medicine. We present our dual-molecular-switch model for Ca2+ regulation of gating functions of voltage-gated sodium channels in which both CaM and an intrinsic EF-hand domain serve as coupled Ca2+ sensors. A second example involves novel EFCaBP extracellular function, that is, the role of S100A8/S100A9 heterodimer in the innate immune response to bacterial pathogens. A mechanism for the antimicrobial activity of S100A8/S100A9 was discovered. We describe interactions of S100A8/S100A9 and S100B with the cell surface receptor for advanced glycation end products. Biochemical and structural studies are now uncovering the mechanisms by which EFCaBPs work and are helping to define their biological activities, while simultaneously expanding knowledge of the roles of these proteins in normal cellular physiology and the pathology of disease.
Co-reporter:Kyle A. Nordquist, Yoana N. Dimitrova, Peter S. Brzovic, Whitney B. Ridenour, Kim A. Munro, Sarah E. Soss, Richard M. Caprioli, Rachel E. Klevit and Walter J. Chazin
Biochemistry 2010 Volume 49(Issue 2) pp:
Publication Date(Web):December 17, 2009
DOI:10.1021/bi901620v
Substantial evidence has accumulated indicating a significant role for oligomerization in the function of E3 ubiquitin ligases. Among the many characterized E3 ligases, the yeast U-box protein Ufd2 and its mammalian homologue E4B appear to be unique in functioning as monomers. An E4B U-box domain construct (E4BU) has been subcloned, overexpressed in Escherichia coli, and purified, which enabled determination of a high-resolution NMR solution structure and detailed biophysical analysis. E4BU is a stable monomeric protein that folds into the same structure observed for other structurally characterized U-box domain homodimers. Multiple sequence alignment combined with comparative structural analysis reveals substitutions in the sequence that inhibit dimerization. The interaction between E4BU and the E2 conjugating enzyme UbcH5c has been mapped using NMR, and these data have been used to generate a structural model for the complex. The E2 binding site is found to be similar to that observed for dimeric U-box and RING domain E3 ligases. Despite the inability to dimerize, E4BU was found to be active in a standard autoubiquitination assay. The structure of E4BU and its ability to function as a monomer are discussed in light of the ubiquitous observation of U-box and RING domain oligomerization.
Co-reporter:Dalyir I. Pretto, Susan Tsutakawa, Chris A. Brosey, Amalchi Castillo, Marie-Eve Chagot, Jarrod A. Smith, John A. Tainer and Walter J. Chazin
Biochemistry 2010 Volume 49(Issue 13) pp:
Publication Date(Web):February 25, 2010
DOI:10.1021/bi9019934
Replication protein A (RPA) is the primary eukaryotic single-stranded DNA (ssDNA) binding protein utilized in diverse DNA transactions in the cell. RPA is a heterotrimeric protein with seven globular domains connected by flexible linkers, which enable substantial interdomain motion that is essential to its function. Small angle X-ray scattering (SAXS) experiments with two multidomain constructs from the N-terminus of the large subunit (RPA70) were used to examine the structural dynamics of these domains and their response to the binding of ssDNA. The SAXS data combined with molecular dynamics simulations reveal substantial interdomain flexibility for both RPA70AB (the tandem high-affinity ssDNA binding domains A and B connected by a 10-residue linker) and RPA70NAB (RPA70AB extended by a 70-residue linker to the RPA70N protein interaction domain). Binding of ssDNA to RPA70NAB reduces the interdomain flexibility between the A and B domains but has no effect on RPA70N. These studies provide the first direct measurements of changes in orientation of these three RPA domains upon binding ssDNA. The results support a model in which RPA70N remains structurally independent of RPA70AB in the DNA-bound state and therefore freely available to serve as a protein recruitment module.
Co-reporter:Sivaraja Vaithiyalingam;Eric M. Warren;Brandt F. Eichman;
Proceedings of the National Academy of Sciences 2010 107(31) pp:13684-13689
Publication Date(Web):July 19, 2010
DOI:10.1073/pnas.1002009107
DNA replication requires priming of DNA templates by enzymes known as primases. Although DNA primase structures are available
from archaea and bacteria, the mechanism of DNA priming in higher eukaryotes remains poorly understood in large part due to
the absence of the structure of the unique, highly conserved C-terminal regulatory domain of the large subunit (p58C). Here,
we present the structure of this domain determined to 1.7-Å resolution by X-ray crystallography. The p58C structure reveals
a novel arrangement of an evolutionarily conserved 4Fe-4S cluster buried deeply within the protein core and is not similar
to any known protein structure. Analysis of the binding of DNA to p58C by fluorescence anisotropy measurements revealed a
strong preference for ss/dsDNA junction substrates. This approach was combined with site-directed mutagenesis to confirm that
the binding of DNA occurs to a distinctively basic surface on p58C. A specific interaction of p58C with the C-terminal domain
of the intermediate subunit of replication protein A (RPA32C) was identified and characterized by isothermal titration calorimetry
and NMR. Restraints from NMR experiments were used to drive computational docking of the two domains and generate a model
of the p58C–RPA32C complex. Together, our results explain functional defects in human DNA primase mutants and provide insights
into primosome loading on RPA-coated ssDNA and regulation of primase activity.
Co-reporter:Chris A. Brosey ; Marie-Eve Chagot ; Mark Ehrhardt ; Dalyir I. Pretto ; Brian E. Weiner
Journal of the American Chemical Society 2009 Volume 131(Issue 18) pp:6346-6347
Publication Date(Web):April 20, 2009
DOI:10.1021/ja9013634
Modular proteins with multiple domains tethered by flexible linkers have variable global architectures. Using the eukaryotic ssDNA binding protein, Replication Protein A (RPA), we demonstrate that NMR spectroscopy is a powerful tool to characterize the remodeling of architecture in different functional states. The first direct evidence is obtained for the remodeling of RPA upon binding ssDNA, including an alteration in the availability of the RPA32N domain that may help explain its damage-dependent phosphorylation.
Co-reporter:Young-Tae Lee, Yoana N. Dimitrova, Gabriela Schneider, Whitney B. Ridenour, Shibani Bhattacharya, Sarah E. Soss, Richard M. Caprioli, Anna Filipek and Walter J. Chazin
Biochemistry 2008 Volume 47(Issue 41) pp:
Publication Date(Web):September 20, 2008
DOI:10.1021/bi801233z
S100A6 is a member of the S100 subfamily of EF-hand Ca2+ binding proteins that has been shown to interact with calcyclin binding protein/Siah-1 interacting protein (CacyBP/SIP or SIP), a subunit of an SCF-like E3 ubiquitin ligase complex (SCF-TBL1) formed under genotoxic stress. SIP serves as a scaffold in this complex, linking the E2-recruiting module Siah-1 to the substrate-recruiting module Skp1-TBL1. A cell-based functional assay suggests that S100A6 modulates the activity of SCF-TBL1. The results from the cell-based experiments could be enhanced if it were possible to selectively inhibit S100A6−SIP interactions without perturbing any other functions of the two proteins. To this end, the structure of the S100A6−SIP complex was determined in solution by NMR and the strength of the interaction was characterized by isothermal titration calorimetry. In an initial step, the minimal S100A6 binding region in SIP was mapped to a 31-residue fragment (Ser189−Arg219) in the C-terminal domain. The structure of the S100A6−SIP(189−219) complex revealed that SIP(189−219) forms two helices, the first of which (Met193−Tyr200) interacts with S100A6 in a canonical binding mode. The second helix (Met207−Val216) lies over the S100A6 dimer interface, a mode of binding to S100A6 that has not previously been observed for any target bound to an S100 protein. A series of structure-based SIP mutations showed reduced S100A6 binding affinity, setting the stage for direct functional analysis of S100A6−SIP interactions.
Co-reporter:Vikas N. Shah;Tammy L. Wingo;Kevin L. Weiss;Christina K. Williams;Jeffrey R. Balser;
Proceedings of the National Academy of Sciences 2006 103(10) pp:3592-3597
Publication Date(Web):February 27, 2006
DOI:10.1073/pnas.0507397103
The function of the human cardiac voltage-gated sodium channel NaV1.5 (hH1) is regulated in part by binding of calcium to an EF hand in the C-terminal cytoplasmic domain. hH1 is also regulated
via an extrinsic calcium-sensing pathway mediated by calmodulin (CaM) via binding to an IQ motif immediately adjacent to the
EF-hand domain. The intrinsic EF-hand domain is shown here to interact with the IQ motif, which controls calcium affinity.
Remarkably, mutation of the IQ residues has only a minor effect on CaM affinity but drastically reduces calcium affinity of
the EF-hand domain, whereas the Brugada mutation A1924T significantly reduces CaM affinity but has no effect on calcium affinity
of the EF-hand domain. Moreover, the differences in the biochemical effects of the mutations directly correlate with contrasting
effects on channel electrophysiology. A comprehensive model is proposed in which the hH1 IQ motif serves as a molecular switch,
coupling the intrinsic and extrinsic calcium sensors.
Co-reporter:
Nature Structural and Molecular Biology 2005 12(4) pp:332-339
Publication Date(Web):27 March 2005
DOI:10.1038/nsmb916
Simian virus 40 (SV40) provides a model system for the study of eukaryotic DNA replication, in which the viral protein, large T antigen (Tag), marshals human proteins to replicate the viral minichromosome. SV40 replication requires interaction of Tag with the host single-stranded DNA-binding protein, replication protein A (hRPA). The C-terminal domain of the hRPA32 subunit (RPA32C) facilitates initiation of replication, but whether it interacts with Tag is not known. Affinity chromatography and NMR revealed physical interaction between hRPA32C and the Tag origin DNA−binding domain, and a structural model of the complex was determined. Point mutations were then designed to reverse charges in the binding sites, resulting in substantially reduced binding affinity. Corresponding mutations introduced into intact hRPA impaired initiation of replication and primosome activity, implying that this interaction has a critical role in assembly and progression of the SV40 replisome.
Co-reporter:
Nature Structural and Molecular Biology 2004 11(3) pp:219-225
Publication Date(Web):22 February 2004
DOI:10.1038/nsmb737
Sodium channels initiate the electrical cascade responsible for cardiac rhythm, and certain life-threatening arrhythmias arise from Na+ channel dysfunction. We propose a novel mechanism for modulation of Na+ channel function whereby calcium ions bind directly to the human cardiac Na+ channel (hH1) via an EF-hand motif in the C-terminal domain. A functional role for Ca2+ binding was identified electrophysiologically, by measuring Ca2+-induced modulation of hH1. A small hH1 fragment containing the EF-hand motif was shown to form a structured domain and to bind Ca2+ with affinity characteristic of calcium sensor proteins. Mutations in this domain reduce Ca2+ affinity in vitro and the inactivation gating effects of Ca2+ in electrophysiology experiments. These studies reveal the molecular basis for certain forms of long QT syndrome and other arrhythmia-producing syndromes, and suggest a potential pharmacological target for antiarrhythmic drug design.
Co-reporter:
Nature Structural and Molecular Biology 2003 10(4) pp:250-255
Publication Date(Web):03 March 2003
DOI:10.1038/nsb906
The structure of the U-box in the essential Saccharomyces cerevisiae pre-mRNA splicing factor Prp19p has been determined by NMR. The conserved zinc-binding sites supporting the cross-brace arrangement in RING-finger domains are replaced by hydrogen-bonding networks in the U-box. These hydrogen-bonding networks are necessary for the structural stabilization and activity of the U-box. A conservative ValIle point mutation in the Prp19p U-box domain leads to pre-mRNA splicing defects in vivo. NMR analysis of this mutant shows that the substitution disrupts structural integrity of the U-box domain. Furthermore, comparison of the Prp19p U-box domain with known RING−E2 complex structures demonstrates that both U-box and RING-fingers contain a conserved interaction surface. Mutagenesis of residues at this interface, while not perturbing the structure of the U-box, abrogates Prp19p function in vivo. These comparative structural and functional analyses imply that the U-box and its associated ubiquitin ligase activity are critical for Prp19p function in vivo.
Co-reporter:
Nature Structural and Molecular Biology 2001 8(11) pp:910 - 912
Publication Date(Web):
DOI:10.1038/nsb1101-910
Co-reporter:
Nature Structural and Molecular Biology 2000 7(3) pp:245 - 250
Publication Date(Web):
DOI:10.1038/73369
Co-reporter:Steven M. Shell, Edward K. Hawkins, Miaw-Sheue Tsai, Aye Su Hlaing, Carmelo J. Rizzo, Walter J. Chazin
DNA Repair (November 2013) Volume 12(Issue 11) pp:947-953
Publication Date(Web):1 November 2013
DOI:10.1016/j.dnarep.2013.08.013
•High-throughput fluorescence anisotropy assay used to measure affinity of human XPC for damaged DNA.•Binding affinity tested for eight DNA lesions contained in a common DNA sequence.•XPC binds all damaged DNA with essentially the same affinity.•XPC efficiently recognizes oxidative lesions repaired by BER pathway.•XPC may coordinate recognition of damaged DNA for multiple repair pathways.The Xeroderma pigmentosum complementation group C protein (XPC) serves as the primary initiating factor in the global genome nucleotide excision repair pathway (GG-NER). Recent reports suggest XPC also stimulates repair of oxidative lesions by base excision repair. However, whether XPC distinguishes among various types of DNA lesions remains unclear. Although the DNA binding properties of XPC have been studied by several groups, there is a lack of consensus over whether XPC discriminates between DNA damaged by lesions associated with NER activity versus those that are not. In this study we report a high-throughput fluorescence anisotropy assay used to measure the DNA binding affinity of XPC for a panel of DNA substrates containing a range of chemical lesions in a common sequence. Our results demonstrate that while XPC displays a preference for binding damaged DNA, the identity of the lesion has little effect on the binding affinity of XPC. Moreover, XPC was equally capable of binding to DNA substrates containing lesions not repaired by GG-NER. Our results suggest XPC may act as a general sensor of damaged DNA that is capable of recognizing DNA containing lesions not repaired by NER.
Co-reporter:Norie Sugitani, Walter J. Chazin
Progress in Biophysics and Molecular Biology (March 2015) Volume 117(Issues 2–3) pp:206-211
Publication Date(Web):1 March 2015
DOI:10.1016/j.pbiomolbio.2014.12.001
DNA replication, damage response and repair require the coordinated action of multi-domain proteins operating within dynamic multi-protein machines that act upon the DNA substrate. These modular proteins contain flexible linkers of various lengths, which enable changes in the spatial distribution of the globular domains (architecture) that harbor their essential biochemical functions. This mobile architecture is uniquely suited to follow the evolving substrate landscape present over the course of the specific process performed by the multi-protein machinery. A fundamental advance in understanding of protein machinery is the realization of the pervasive role of dynamics. Not only is the machine undergoing dynamic transformations, but the proteins themselves are flexible and constantly adapting to the progression through the steps of the overall process. Within this dynamic context the activity of the constituent proteins must be coordinated, a role typically played by hub proteins. A number of important characteristics of modular proteins and concepts about the operation of dynamic machinery have been discerned. These provide the underlying basis for the action of the machinery that reads DNA, and responds to and repairs DNA damage. Here, we introduce a number of key characteristics and concepts, including the modularity of the proteins, linkage of weak binding sites, direct competition between sites, and allostery, using the well recognized hub protein replication protein A (RPA).
Co-reporter:Walter J. Chazin
Structure (January 2008) Volume 16(Issue 1) pp:12-14
Publication Date(Web):1 January 2008
DOI:10.1016/j.str.2007.11.002
Co-reporter:Steven M. Shell, Walter J. Chazin
Structure (4 April 2012) Volume 20(Issue 4) pp:566-568
Publication Date(Web):4 April 2012
DOI:10.1016/j.str.2012.03.004
In this issue of Structure, Das et al. report the structure of the helix-hairpin-helix dimerization domain of XPF bound to ssDNA. These results provide insight into the architecture of nucleotide excision repair machinery and how it interacts with damaged DNA substrates.
Co-reporter:Chris A. Brosey, Sarah E. Soss, Sonja Brooks, Chunli Yan, ... Walter J. Chazin
Structure (2 June 2015) Volume 23(Issue 6) pp:1028-1038
Publication Date(Web):2 June 2015
DOI:10.1016/j.str.2015.04.008
•DNA binding dramatically reorients and couples the inter-domain motion of RPA70AB•RPA70N protein interaction domain has structural and dynamic autonomy from RPA70AB•RPA70N remains autonomous from RPA70AB when ssDNA is engaged•Linkers between the globular domains are proposed to control RPA functional dynamicsReplication Protein A (RPA) is an essential scaffold for many DNA processing machines; its function relies on its modular architecture. Here, we report 15N-nuclear magnetic resonance heteronuclear relaxation analysis to characterize the movements of single-stranded (ss) DNA binding and protein interaction modules in the RPA70 subunit. Our results provide direct evidence for coordination of the motion of the tandem RPA70AB ssDNA binding domains. Moreover, binding of ssDNA substrate is found to cause dramatic reorientation and full coupling of inter-domain motion. In contrast, the RPA70N protein interaction domain remains structurally and dynamically independent of RPA70AB regardless of binding of ssDNA. This autonomy of motion between the 70N and 70AB modules supports a model in which the two binding functions of RPA are mediated fully independently, but remain differentially coordinated depending on the length of their flexible tethers. A critical role for linkers between the globular domains in determining the functional dynamics of RPA is proposed.Download high-res image (227KB)Download full-size image
Co-reporter:Weixing Zhao, Sivaraja Vaithiyalingam, Joseph San Filippo, David G. Maranon, ... Patrick Sung
Molecular Cell (16 July 2015) Volume 59(Issue 2) pp:176-187
Publication Date(Web):16 July 2015
DOI:10.1016/j.molcel.2015.05.032
•DSS1 works in conjunction with BRCA2 to facilitate RPA-RAD51 exchange on ssDNA•DSS1 interacts with RPA via its solvent-exposed acidic loop•DSS1 binds the RPA70 subunit of RPA and attenuates ssDNA binding by it•DSS1 could also function as a nucleic acid mimic in other biological processesThe tumor suppressor BRCA2 is thought to facilitate the handoff of ssDNA from replication protein A (RPA) to the RAD51 recombinase during DNA break and replication fork repair by homologous recombination. However, we find that RPA-RAD51 exchange requires the BRCA2 partner DSS1. Biochemical, structural, and in vivo analyses reveal that DSS1 allows the BRCA2-DSS1 complex to physically and functionally interact with RPA. Mechanistically, DSS1 acts as a DNA mimic to attenuate the affinity of RPA for ssDNA. A mutation in the solvent-exposed acidic domain of DSS1 compromises the efficacy of RPA-RAD51 exchange. Thus, by targeting RPA and mimicking DNA, DSS1 functions with BRCA2 in a two-component homologous recombination mediator complex in genome maintenance and tumor suppression. Our findings may provide a paradigm for understanding the roles of DSS1 in other biological processes.Download high-res image (157KB)Download full-size image
Co-reporter:Christina K. Williams, Sivaraja Vaithiyalingam, Michal Hammel, James Pipas, Walter J. Chazin
Archives of Biochemistry and Biophysics (15 February 2012) Volume 518(Issue 2) pp:
Publication Date(Web):15 February 2012
DOI:10.1016/j.abb.2011.12.014
Simian Virus 40 uses the large T antigen (Tag) to bind and inactivate retinoblastoma tumor suppressor proteins (Rb), which can result in cellular transformation. Tag is a modular protein with four domains connected by flexible linkers. The N-terminal J domain of Tag is necessary for Rb inactivation. Binding of Rb is mediated by an LXCXE consensus motif immediately C-terminal to the J domain. Nuclear magnetic resonance (NMR) and small angle X-ray scattering (SAXS) were used to study the structural dynamics and interaction of Rb with the LXCXE motif, the J domain and a construct (N260) extending from the J domain through the origin binding domain (OBD). NMR and SAXS data revealed substantial flexibility between the domains in N260. Binding of pRb to a construct containing the LXCXE motif and the J domain revealed weak interactions between pRb and the J domain. Analysis of the complex of pRb and N260 indicated that the OBD is not involved and retains its dynamic independence from the remainder of Tag. These results support a ‘chaperone’ model in which the J domain of Tag changes its orientation as it acts upon different protein complexes.Highlights► The data reveal flexible linkage of origin binding and J domains of SV40 T antigen. ► Contacts between J domain and pRb in the crystal structure are transient in solution. ► The origin binding domain remains dynamically independent in the complex with pRb. ► The results support a ‘chaperone’ model for the action of the J domain.
Co-reporter:Michael Koch, Seth Chitayat, Brian M. Dattilo, Andre Schiefner, ... Günter Fritz
Structure (13 October 2010) Volume 18(Issue 10) pp:1342-1352
Publication Date(Web):13 October 2010
DOI:10.1016/j.str.2010.05.017
The receptor for advanced glycation end products (RAGE) is a pattern recognition receptor involved in inflammatory processes and is associated with diabetic complications, tumor outgrowth, and neurodegenerative disorders. RAGE induces cellular signaling events upon binding of a variety of ligands, such as glycated proteins, amyloid-β, HMGB1, and S100 proteins. The X-ray crystal structure of the VC1 ligand-binding region of the human RAGE ectodomain was determined at 1.85 Å resolution. The VC1 ligand-binding surface was mapped onto the structure from titrations with S100B monitored by heteronuclear NMR spectroscopy. These NMR chemical shift perturbations were used as input for restrained docking calculations to generate a model for the VC1-S100B complex. Together, the arrangement of VC1 molecules in the crystal and complementary biochemical studies suggest a role for self-association in RAGE function. Our results enhance understanding of the functional outcomes of S100 protein binding to RAGE and provide insight into mechanistic models for how the receptor is activated.Graphical AbstractDownload high-res image (478KB)Download full-size image
Co-reporter:Benjamin Chagot, Walter J. Chazin
Journal of Molecular Biology (11 February 2011) Volume 406(Issue 1) pp:106-119
Publication Date(Web):11 February 2011
DOI:10.1016/j.jmb.2010.11.046
The function of the human voltage-gated sodium channel NaV1.5 is regulated in part by intracellular calcium signals. The ubiquitous calcium sensor protein calmodulin (CaM) is an important part of the complex calcium-sensing apparatus in NaV1.5. CaM interacts with an IQ (isoleucine–glutamine) motif in the large intracellular C-terminal domain of the channel. Using co-expression and co-purification, we have been able to isolate a CaM–IQ motif complex and to determine its high-resolution structure in absence of calcium using multi-dimensional solution NMR. Under these conditions, the NaV1.5 IQ motif interacts with the C-terminal domain (C-lobe) of CaM, with the N-terminal domain remaining free in solution. The structure reveals that the C-lobe adopts a semi-open conformation with the IQ motif bound in a narrow hydrophobic groove. Sequence similarities between voltage-gated sodium channels and voltage-gated calcium channels suggest that the structure of the CaM–NaV1.5 IQ motif complex can serve as a general model for the interaction between CaM and ion channel IQ motifs under low-calcium conditions. The structure also provides insight into the biochemical basis for disease-associated mutations that map to the IQ motif in NaV1.5.Download high-res image (300KB)Download full-size imageResearch Highlights► The first structure of apo-CaM bound to an ion channel IQ motif is presented. ► Apo-CaM binds the NaV1.5 IQ motif using only its C-terminal domain. ► The CaM C-terminal domain occupies a semi-open conformation. ► This structure serves as a general model for Apo-CaM–ion channel IQ motif complexes. ► Structure suggests that the NaV1.5 long QT syndrome mutant S1904L is defective in CaM binding.
.jpg)

























































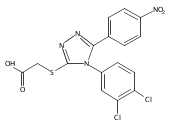
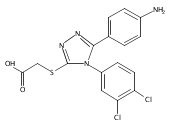
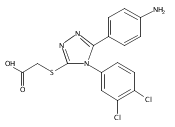
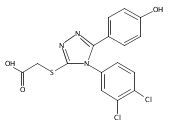
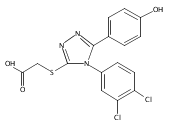
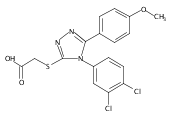
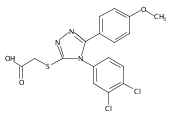
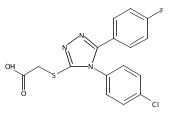

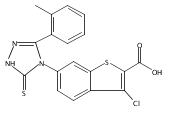
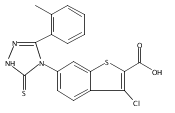

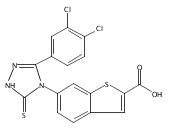
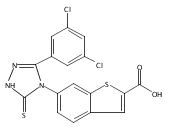








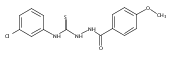
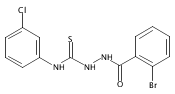
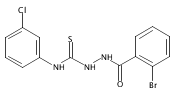
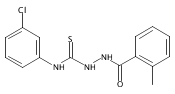


![6-Amino-3-chloro-benzo[b]thiophene-2-carboxylic acid methyl ester](http://img.cochemist.com/ccimg/832800/832739-91-0.png)
![6-Amino-3-chloro-benzo[b]thiophene-2-carboxylic acid methyl ester](http://img.cochemist.com/ccimg/832800/832739-91-0_b.png)


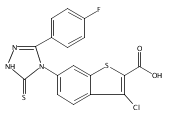

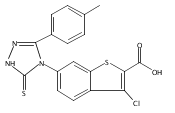
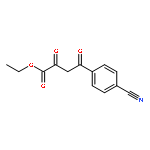
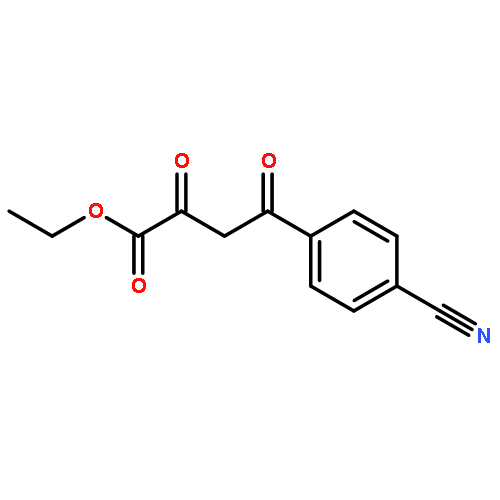
![4-(3,4-Dichloro-phenyl)-5-(4-methoxy-phenyl)-4H-[1,2,4]triazole-3-thiol](http://img.cochemist.com/ccimg/438100/438031-65-3.png)
![4-(3,4-Dichloro-phenyl)-5-(4-methoxy-phenyl)-4H-[1,2,4]triazole-3-thiol](http://img.cochemist.com/ccimg/438100/438031-65-3_b.png)


![3-Chloro-6-methylbenzo[b]thiophene-2-carbohydrazide](http://img.cochemist.com/ccimg/355900/355815-78-0.png)
![3-Chloro-6-methylbenzo[b]thiophene-2-carbohydrazide](http://img.cochemist.com/ccimg/355900/355815-78-0_b.png)





![Benzo[b]thiophene-2-carbohydrazide](http://img.cochemist.com/ccimg/175200/175135-07-6.png)
![Benzo[b]thiophene-2-carbohydrazide](http://img.cochemist.com/ccimg/175200/175135-07-6_b.png)




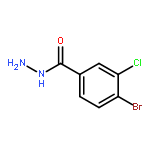
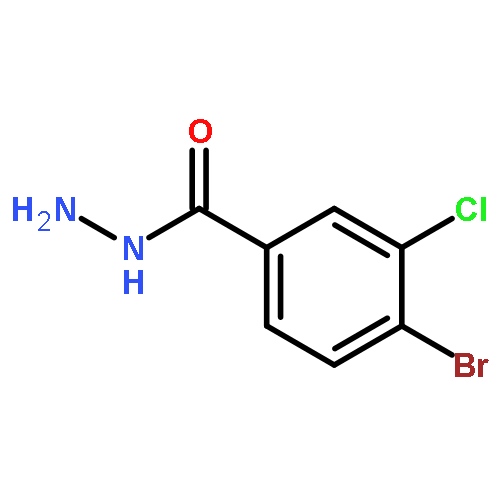
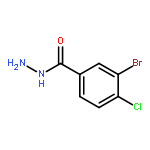
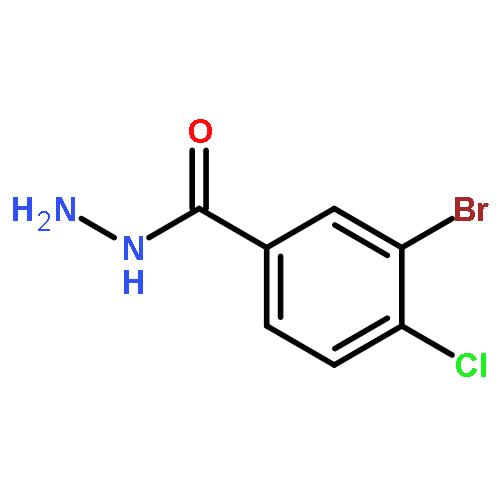
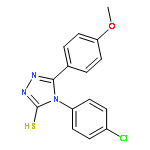
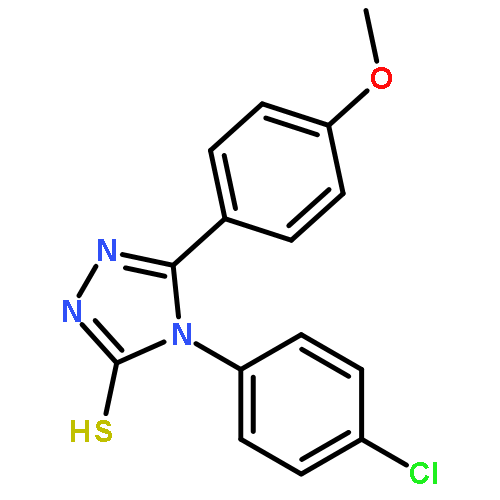


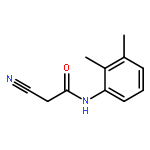



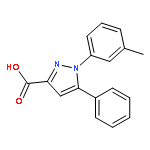
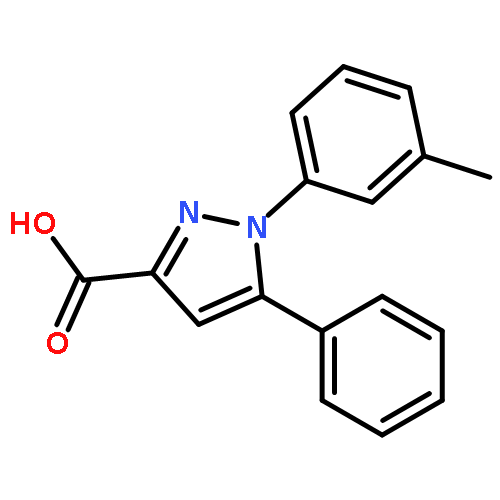
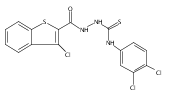
![Benzo[b]thiophene-2-carboxylic acid, 3-chloro-6-nitro-, methyl ester](http://img.cochemist.com/ccimg/59900/59812-36-1.png)
![Benzo[b]thiophene-2-carboxylic acid, 3-chloro-6-nitro-, methyl ester](http://img.cochemist.com/ccimg/59900/59812-36-1_b.png)
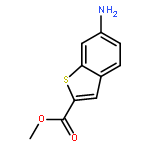
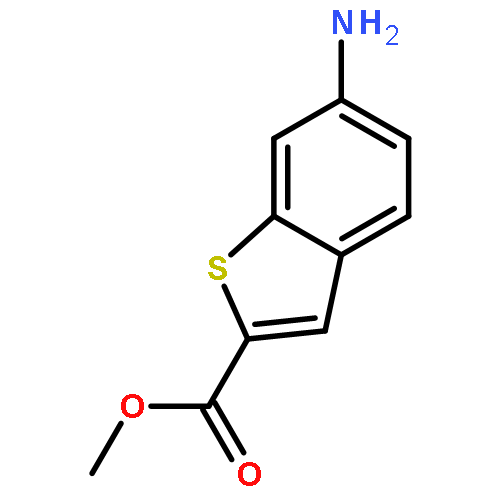
![PYRIMIDINE, 1,4,5,6-TETRAHYDRO-1-METHYL-2-[2-(2-THIENYL)ETHENYL]-](http://img.cochemist.com/ccimg/5700/5686-02-2.png)
![PYRIMIDINE, 1,4,5,6-TETRAHYDRO-1-METHYL-2-[2-(2-THIENYL)ETHENYL]-](http://img.cochemist.com/ccimg/5700/5686-02-2_b.png)