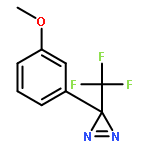
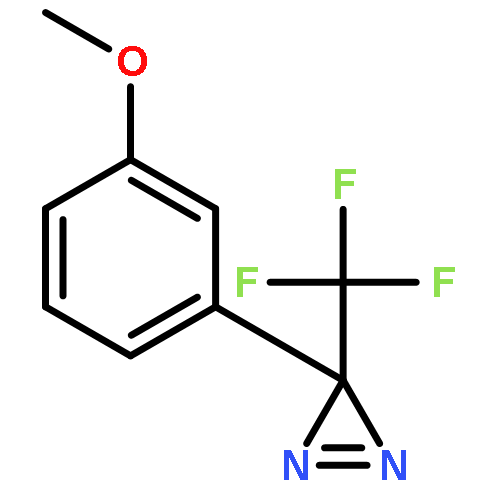
Co-reporter:Elliot J. Lawrence, Ewan R. Clark, Liam D. Curless, James M. Courtney, Robin J. Blagg, Michael J. Ingleson and Gregory G. Wildgoose
Chemical Science 2016 vol. 7(Issue 4) pp:2537-2543
Publication Date(Web):06 Jan 2016
DOI:10.1039/C5SC04564A
Whilst hydrogen is a potentially clean fuel for energy storage and utilisation technologies, its conversion to electricity comes at a high energetic cost. This demands the use of rare and expensive precious metal electrocatalysts. Electrochemical-frustrated Lewis pairs offer a metal-free, CO tolerant pathway to the electrocatalysis of hydrogen oxidation. They function by combining the hydrogen-activating ability of frustrated Lewis pairs (FLPs) with electrochemical oxidation of the resultant hydride. Here we present an electrochemical–FLP approach that utilises two different Lewis acids – a carbon-based N-methylacridinium cation that possesses excellent electrochemical attributes, and a borane that exhibits fast hydrogen cleavage kinetics and functions as a “hydride shuttle”. This synergistic interaction provides a system that is electrocatalytic with respect to the carbon-based Lewis acid, decreases the required potential for hydrogen oxidation by 1 V, and can be recycled multiple times.
Co-reporter:Robin J. Blagg, Trevor R. Simmons, Georgina R. Hatton, James M. Courtney, Elliot L. Bennett, Elliot J. Lawrence and Gregory G. Wildgoose
Dalton Transactions 2016 vol. 45(Issue 14) pp:6032-6043
Publication Date(Web):30 Oct 2015
DOI:10.1039/C5DT03854E
A series of homo- and hetero-tri(aryl)boranes incorporating pentafluorophenyl, 3,5-bis(trifluoromethyl)phenyl, and pentachlorophenyl groups, four of which are novel species, have been studied as the acidic component of frustrated Lewis pairs for the heterolytic cleavage of H2. Under mild conditions eight of these will cleave H2; the rate of cleavage depending on both the electrophilicity of the borane and the steric bulk around the boron atom. Electrochemical studies allow comparisons of the electrophilicity with spectroscopic measurements of Lewis acidity for different series of boranes. Discrepancies in the correlation between these two types of measurements, combined with structural characterisation of each borane, reveal that the twist of the aryl rings with respect to the boron-centred trigonal plane is significant from both a steric and electronic perspective, and is an important consideration in the design of tri(aryl)boranes as Lewis acids.
Co-reporter:Robin J. Blagg, Elliot J. Lawrence, Katie Resner, Vasily S. Oganesyan, Thomas J. Herrington, Andrew E. Ashley and Gregory G. Wildgoose
Dalton Transactions 2016 vol. 45(Issue 14) pp:6023-6031
Publication Date(Web):28 Jul 2015
DOI:10.1039/C5DT01918D
Three structural isomers of tris{bis(trifluoromethyl)phenyl}borane have been studied as the acidic component of frustrated Lewis pairs. While the 3,5-substituted isomer is already known to heterolytically cleave H2 to generate a bridging-hydride; ortho-substituents in the 2,4- and 2,5-isomers quench such reactivity through electron donation into the vacant boron pz orbital and steric blocking of the boron centre; as shown by electrochemical, structural and computational studies. Electrochemical studies of the corresponding borohydrides identify that the two-electron oxidation of terminal-hydrides occurs at more positive potentials than observed for [HB(C6F5)3]−, while the bridging-hydride oxidizes at a higher potential still, comparable to that of free H2.
Co-reporter:Robin J. Blagg and Gregory G. Wildgoose
RSC Advances 2016 vol. 6(Issue 48) pp:42421-42427
Publication Date(Web):28 Apr 2016
DOI:10.1039/C6RA07007H
The novel 1:1:1 hetero-tri(aryl)borane (pentafluorophenyl){3,5-bis(trifluoromethyl)phenyl}(pentachlorophenyl)borane has been synthesised and structurally characterised. This has been show to act as the Lewis acidic component in FLPs for the heterolytic cleavage of H2 with three Lewis bases.
Co-reporter:James P. Buttress, David P. Day, James M. Courtney, Elliot J. Lawrence, David L. Hughes, Robin J. Blagg, Alison Crossley, Susan E. Matthews, Carl Redshaw, Philip C. Bulman Page, and Gregory G. Wildgoose
Langmuir 2016 Volume 32(Issue 31) pp:7806-7813
Publication Date(Web):July 15, 2016
DOI:10.1021/acs.langmuir.6b02222
We herein report the synthesis of novel “Janus” calix[4]arenes bearing four “molecular tethering” functional groups on either the upper or lower rims of the calixarene. These enable facile multipoint covalent attachment to electrode surfaces with monolayer coverage. The other rim of the calixarenes bear either four azide or four ethynyl functional groups, which are easily modified by the copper(I)-catalyzed azide–alkyne cycloaddition reaction (CuAAC), either pre- or postsurface modification, enabling these conical, nanocavity reactor sites to be decorated with a wide range of substrates to impart desired chemical properties. Redox active species decorating the peripheral rim are shown to be electrically connected by the calixarene to the electrode surface in either “up” or “down” orientations of the calixarene.
Co-reporter:M. Concepción Gimeno; José M. López-de-Luzuriaga; Elena Manso; Miguel Monge; M. Elena Olmos; María Rodríguez-Castillo; María-Teresa Tena; David P. Day; Elliot J. Lawrence
Inorganic Chemistry 2015 Volume 54(Issue 22) pp:10667-10677
Publication Date(Web):October 23, 2015
DOI:10.1021/acs.inorgchem.5b01477
Reaction of [Au(C6F5)(tht)] (tht = tetrahydrothiophene) with 2,2′:6′,2″-terpyridine (terpy) leads to complex [Au(C6F5)(η1-terpy)] (1). The chemical oxidation of complex (1) with 2 equiv of [N(C6H4Br-4)3](PF6) or using electrosynthetic techniques affords the Au(III) complex [Au(C6F5)(η3-terpy)](PF6)2 (2). The X-ray diffraction study of complex 2 reveals that the terpyridine acts as tridentate chelate ligand, which leads to a slightly distorted square-planar geometry. Complex 1 displays fluorescence in the solid state at 77 K due to a metal (gold) to ligand (terpy) charge transfer transition, whereas complex 2 displays fluorescence in acetonitrile due to excimer or exciplex formation. Time-dependent density functional theory calculations match the experimental absorption spectra of the synthesized complexes. In order to further probe the frontier orbitals of both complexes and study their redox behavior, each compound was separately characterized using cyclic voltammetry. The bulk electrolysis of a solution of complex 1 was analyzed by spectroscopic methods confirming the electrochemical synthesis of complex 2.
Co-reporter:Elliot J. Lawrence;Dr. Robin J. Blagg;Dr. David L. Hughes;Dr. Andrew E. Ashley;Dr. Gregory G. Wildgoose
Chemistry - A European Journal 2015 Volume 21( Issue 2) pp:900-906
Publication Date(Web):
DOI:10.1002/chem.201404242
Abstract
Herein, we extend our “combined electrochemical–frustrated Lewis pair” approach to include Pt electrode surfaces for the first time. We found that the voltammetric response of an electrochemical–frustrated Lewis pair (FLP) system involving the B(C6F5)3/[HB(C6F5)3]− redox couple exhibits a strong surface electrocatalytic effect at Pt electrodes. Using a combination of kinetic competition studies in the presence of a H atom scavenger, 6-bromohexene, and by changing the steric bulk of the Lewis acid borane catalyst from B(C6F5)3 to B(C6Cl5)3, the mechanism of electrochemical–FLP reactions on Pt surfaces was shown to be dominated by hydrogen-atom transfer (HAT) between Pt, [PtH] adatoms and transient [HB(C6F5)3]⋅ electrooxidation intermediates. These findings provide further insight into this new area of combining electrochemical and FLP reactions, and proffers additional avenues for exploration beyond energy generation, such as in electrosynthesis.
Co-reporter:Elliot J. Lawrence ; Vasily S. Oganesyan ; David L. Hughes ; Andrew E. Ashley
Journal of the American Chemical Society 2014 Volume 136(Issue 16) pp:6031-6036
Publication Date(Web):April 2, 2014
DOI:10.1021/ja500477g
Frustrated Lewis pairs have found many applications in the heterolytic activation of H2 and subsequent hydrogenation of small molecules through delivery of the resulting proton and hydride equivalents. Herein, we describe how H2 can be preactivated using classical frustrated Lewis pair chemistry and combined with in situ nonaqueous electrochemical oxidation of the resulting borohydride. Our approach allows hydrogen to be cleanly converted into two protons and two electrons in situ, and reduces the potential (the required energetic driving force) for nonaqueous H2 oxidation by 610 mV (117.7 kJ mol–1). This significant energy reduction opens routes to the development of nonaqueous hydrogen energy technology.
Co-reporter:David P. Day, Thomas Dann, David. L. Hughes, Vasily S. Oganesyan, Dietmar Steverding, and Gregory G. Wildgoose
Organometallics 2014 Volume 33(Issue 18) pp:4687-4696
Publication Date(Web):January 15, 2014
DOI:10.1021/om4007642
We report the first known examples of triazole-derivatized cymantrene complexes (η5-[4-substituted triazol-1-yl]cyclopentadienyl)tricarbonylmanganese(I), obtained via a “click” chemical synthesis, bearing a phenyl, 3-aminophenyl, or 4-aminophenyl moiety at the 4-position of the triazole ring. Structural characterization data using multinuclear NMR, UV–vis, ATR-IR, and mass spectrometric methods are provided, as well as crystallographic data for (η5-[4-phenyltriazol-1-yl]cyclopentadienyl)tricarbonylmanganese(I) and (η5-[4-(3-aminophenyl)triazol-1-yl]cyclopentadienyl)tricarbonylmanganese(I). Cyclic voltammetric characterization of the redox behavior of each of the three cymantrene–triazole complexes is presented together with digital simulations, in situ infrared spectroelectrochemistry, and DFT calculations to extract the associated kinetic and thermodynamic parameters. The trypanocidal activity of each cymantrene–triazole complex is also examined, and these complexes are found to be more active than cymantrene alone.
Co-reporter:Elliot J. Lawrence;Thomas J. Herrington;Dr. Andrew E. Ashley;Dr. Gregory G. Wildgoose
Angewandte Chemie International Edition 2014 Volume 53( Issue 37) pp:9922-9925
Publication Date(Web):
DOI:10.1002/anie.201405721
Abstract
In order to use H2 as a clean source of electricity, prohibitively rare and expensive precious metal electrocatalysts, such as Pt, are often used to overcome the large oxidative voltage required to convert H2 into 2 H+ and 2 e−. Herein, we report a metal-free approach to catalyze the oxidation of H2 by combining the ability of frustrated Lewis pairs (FLPs) to heterolytically cleave H2 with the in situ electrochemical oxidation of the resulting borohydride. The use of the NHC-stabilized borenium cation [(IiPr2)(BC8H14)]+ (IiPr2=C3H2(NiPr)2, NHC=N-heterocyclic carbene) as the Lewis acidic component of the FLP is shown to decrease the voltage required for H2 oxidation by 910 mV at inexpensive carbon electrodes, a significant energy saving equivalent to 175.6 kJ mol−1. The NHC–borenium Lewis acid also offers improved catalyst recyclability and chemical stability compared to B(C6F5)3, the paradigm Lewis acid originally used to pioneer our combined electrochemical/frustrated Lewis pair approach.
Co-reporter:David P. Day, Thomas Dann, Robin J. Blagg, Gregory G. Wildgoose
Journal of Organometallic Chemistry 2014 770() pp: 29-34
Publication Date(Web):
DOI:10.1016/j.jorganchem.2014.07.020
Co-reporter:Elliot J. Lawrence, Vasily S. Oganesyan, Gregory G. Wildgoose and Andrew E. Ashley
Dalton Transactions 2013 vol. 42(Issue 3) pp:782-789
Publication Date(Web):22 Nov 2012
DOI:10.1039/C2DT31622F
We report a kinetic and mechanistic study into the one-electron reduction of the archetypal Lewis acid tris(pentafluorophenyl)borane, B(C6F5)3, in dichloromethane and 1,2-difluorobenzene. Electrochemical experiments, combined with digital simulations, DFT computational studies and multinuclear NMR analysis allow us to obtain thermodynamic, kinetic and mechanistic information relating to the redox activity of B(C6F5)3. We show that tris(pentafluorophenyl)borane undergoes a quasi-reversible one-electron reduction followed by rapid chemical decomposition of the B(C6F5)3˙− radical anion intermediate via a solvolytic radical pathway. The reaction products form various four-coordinate borates of which [B(C6F5)4]− is a very minor product. The rate of the follow-up chemical step has a pseudo-first order rate constant of the order of 6 s−1. This value is three orders of magnitude larger than that found in previous studies performed in the donor solvent, tetrahydrofuran. The standard reduction potential of B(C6F5)3 is reported for the first time as −1.79 ± 0.1 V and −1.65 ± 0.1 V vs. ferrocene/ferrocenium in dichloromethane and 1,2-difluorobenzene respectively.
Co-reporter:Joseph C. Bear, Paul D. McNaughter, Kerstin Jurkschat, Alison Crossley, Leigh Aldous, Richard G. Compton, Andrew G. Mayes, Gregory G. Wildgoose
Journal of Colloid and Interface Science 2012 Volume 383(Issue 1) pp:110-117
Publication Date(Web):1 October 2012
DOI:10.1016/j.jcis.2012.06.028
Herein, we report the synthesis of three covalently linked superparamagnetic nanocrystal-multi-walled carbon nanotube (MWCNT) composites. A generic strategy for amphiphilic polymer coating of nanocrystals and further functionalization for amide bond formation with the MWCNTs is discussed. This approach can in principle allow attachment of any colloidal nanocrystal to the MWCNTs. The materials were characterized at each stage of the syntheses using DLS, zeta-potential measurements, FT-IR, TEM, and XPS techniques. The practicality of this linkage is demonstrated by the reversible magnetic immobilization of these materials on an electrode during non-aqueous electrochemistry.Graphical abstractHighlights► Preparation of superparamagnetic nanoparticles-carbon nanotube composites. ► Polymer coating the nanoparticles passivates them and allows regioselective attachment to CNTs. ► The majority of CNT surface remains available for further modification. ► “Magnetic nanotube” composites are characterized using a variety of techniques.
Co-reporter:Elliot J. Lawrence, Gregory G. Wildgoose, Leigh Aldous, Yimin A. Wu, Jamie H. Warner, Richard G. Compton, and Paul D. McNaughter
Chemistry of Materials 2011 Volume 23(Issue 16) pp:3740
Publication Date(Web):July 26, 2011
DOI:10.1021/cm201461w
3-Aryl-3-(trifluoromethyl)diazirines are shown to be synthetically useful photoactivated carbene precursors that can be used as molecular “tethers” to facilitate the improved covalent surface modification of graphitic carbon and carbon nanotubes with a potentially large variety of chemical species. Proof-of-concept is demonstrated by the synthesis, as well as spectroscopic and electrochemical characterization, followed by photoactivated attachment of the organometallic diazirine derivative, 3-[3-(trifluoromethyl)diazirin-3-yl]phenyl ferrocene monocarboxylate, to the surface of vitreous carbon, and also to two different morphologies of multiwalled carbon nanotubes (“bamboo-like” and “hollow-tube”, denoted as b-MWCNTs and h-MWCNTs, respectively). The latter differ only in the relative amounts of “edge-plane-like” defect sites (at the termini of the nanotubes) and “basal-plane-like” pristine sidewall regions. The facile covalent coupling of the ferrocenyl “probe” moiety to the diazirine “linker” was confirmed by UV–vis, 1H and 19F NMR spectroscopy, and cyclic voltammetry (CV). Upon exposure to UV irradiation in the presence of graphitic materials, the resulting covalent surface attachment of the ferrocenyl groups via the diazirine “linker” was characterized by Raman and X-ray photoelectron spectroscopy (XPS) and by CV experiments performed in nonaqueous electrolyte. The surface coverage of 3-[3-(trifluoromethyl)diazirin-3-yl]phenyl ferrocene monocarboxylate, analyzed from both CV and XPS experiments was found to be 7%–11% of that estimated for a complete monolayer, and was 20-fold greater than that achieved in control experiments that employed conventional covalent modification strategies to form esters between ferrocene methanol and surface carboxylate groups on the graphitic materials. The surface loading of ferrocene groups on the b-MWCNTs was found to be only ca. 60%–70% that achieved on h-MWCNTs, reflecting the ability of the functionalized carbene intermediate formed upon photolysis of the parent diazirine to insert into C═C bonds in the otherwise relatively inert sidewalls of the nanotubes. This was further confirmed by Raman spectroscopic characterization, which revealed that the h-MWCNTs experienced significantly more sidewall functionalization than the b-MWCNTs, yet still retained good electronic conduction in electrochemical experiments. The relative chemical stability of 3-aryl-3-(trifluoromethyl)diazirines, the ease with which they can be potentially be coupled to a large range of different organic, inorganic, and biological species, and the enhanced surface loading that can be achieved as a result of the reactive carbene intermediate formed during their photolysis, render diazirines highly versatile and potent “linker” molecules for the development of chemically modified materials.Keywords: carbene; carbon nanotubes; chemical modification; diazirine; ferrocene-functionalized carbene; photolysis; sidewall functionalization; surface modification; voltammetry;
Co-reporter:Gregory G. Wildgoose, Elliot J. Lawrence, Joseph C. Bear, Paul D. McNaughter
Electrochemistry Communications 2011 Volume 13(Issue 10) pp:1139-1142
Publication Date(Web):October 2011
DOI:10.1016/j.elecom.2011.07.015
Multiwalled carbon nanotubes (MWCNTs) were covalently modified with polymer-coated superparamagnetic Fe3O4 nanoparticles via amide bond formation to surface oxo-groups located predominantly at the ends of the nanotubes to form “magnetic MWCNTs”. The sidewalls of the magnetic MWCNTs were then selectively covalently modified with ferrocenyl groups via the photolysis of 3-[3-(trifluoromethyl) diazirin-3-yl] phenyl ferrocene monocarboxylate, which uses an aryldiazirine moiety as a molecular “tether”. We demonstrate that the assembly of the chemically-modified magnetic MWCNTs onto the surface of a magnetic carbon electrode enables one to obtain stable voltammetric signals of chemically-modified carbon nanotubes in non-aqueous electrolytes even under vigorous hydrodynamic conditions of stirring at 3000 rpm for up to twenty minutes. In contrast, non-magnetic chemically modified MWCNTs are removed from the electrode surface within the first two minutes of stirring.Highlights► Stable non-aqueous voltammetry of chemically modified CNTs achieved. ► CNTs co-modified with superparamagnetic nanoparticles and ferrocenyl groups. ► Selective end and sidewall co-modification.
Co-reporter:Elliot J. Lawrence, Vasily S. Oganesyan, Gregory G. Wildgoose and Andrew E. Ashley
Dalton Transactions 2013 - vol. 42(Issue 3) pp:NaN789-789
Publication Date(Web):2012/11/22
DOI:10.1039/C2DT31622F
We report a kinetic and mechanistic study into the one-electron reduction of the archetypal Lewis acid tris(pentafluorophenyl)borane, B(C6F5)3, in dichloromethane and 1,2-difluorobenzene. Electrochemical experiments, combined with digital simulations, DFT computational studies and multinuclear NMR analysis allow us to obtain thermodynamic, kinetic and mechanistic information relating to the redox activity of B(C6F5)3. We show that tris(pentafluorophenyl)borane undergoes a quasi-reversible one-electron reduction followed by rapid chemical decomposition of the B(C6F5)3˙− radical anion intermediate via a solvolytic radical pathway. The reaction products form various four-coordinate borates of which [B(C6F5)4]− is a very minor product. The rate of the follow-up chemical step has a pseudo-first order rate constant of the order of 6 s−1. This value is three orders of magnitude larger than that found in previous studies performed in the donor solvent, tetrahydrofuran. The standard reduction potential of B(C6F5)3 is reported for the first time as −1.79 ± 0.1 V and −1.65 ± 0.1 V vs. ferrocene/ferrocenium in dichloromethane and 1,2-difluorobenzene respectively.
Co-reporter:Robin J. Blagg, Elliot J. Lawrence, Katie Resner, Vasily S. Oganesyan, Thomas J. Herrington, Andrew E. Ashley and Gregory G. Wildgoose
Dalton Transactions 2016 - vol. 45(Issue 14) pp:NaN6031-6031
Publication Date(Web):2015/07/28
DOI:10.1039/C5DT01918D
Three structural isomers of tris{bis(trifluoromethyl)phenyl}borane have been studied as the acidic component of frustrated Lewis pairs. While the 3,5-substituted isomer is already known to heterolytically cleave H2 to generate a bridging-hydride; ortho-substituents in the 2,4- and 2,5-isomers quench such reactivity through electron donation into the vacant boron pz orbital and steric blocking of the boron centre; as shown by electrochemical, structural and computational studies. Electrochemical studies of the corresponding borohydrides identify that the two-electron oxidation of terminal-hydrides occurs at more positive potentials than observed for [HB(C6F5)3]−, while the bridging-hydride oxidizes at a higher potential still, comparable to that of free H2.
Co-reporter:Robin J. Blagg, Trevor R. Simmons, Georgina R. Hatton, James M. Courtney, Elliot L. Bennett, Elliot J. Lawrence and Gregory G. Wildgoose
Dalton Transactions 2016 - vol. 45(Issue 14) pp:NaN6043-6043
Publication Date(Web):2015/10/30
DOI:10.1039/C5DT03854E
A series of homo- and hetero-tri(aryl)boranes incorporating pentafluorophenyl, 3,5-bis(trifluoromethyl)phenyl, and pentachlorophenyl groups, four of which are novel species, have been studied as the acidic component of frustrated Lewis pairs for the heterolytic cleavage of H2. Under mild conditions eight of these will cleave H2; the rate of cleavage depending on both the electrophilicity of the borane and the steric bulk around the boron atom. Electrochemical studies allow comparisons of the electrophilicity with spectroscopic measurements of Lewis acidity for different series of boranes. Discrepancies in the correlation between these two types of measurements, combined with structural characterisation of each borane, reveal that the twist of the aryl rings with respect to the boron-centred trigonal plane is significant from both a steric and electronic perspective, and is an important consideration in the design of tri(aryl)boranes as Lewis acids.
Co-reporter:Elliot J. Lawrence, Ewan R. Clark, Liam D. Curless, James M. Courtney, Robin J. Blagg, Michael J. Ingleson and Gregory G. Wildgoose
Chemical Science (2010-Present) 2016 - vol. 7(Issue 4) pp:NaN2543-2543
Publication Date(Web):2016/01/06
DOI:10.1039/C5SC04564A
Whilst hydrogen is a potentially clean fuel for energy storage and utilisation technologies, its conversion to electricity comes at a high energetic cost. This demands the use of rare and expensive precious metal electrocatalysts. Electrochemical-frustrated Lewis pairs offer a metal-free, CO tolerant pathway to the electrocatalysis of hydrogen oxidation. They function by combining the hydrogen-activating ability of frustrated Lewis pairs (FLPs) with electrochemical oxidation of the resultant hydride. Here we present an electrochemical–FLP approach that utilises two different Lewis acids – a carbon-based N-methylacridinium cation that possesses excellent electrochemical attributes, and a borane that exhibits fast hydrogen cleavage kinetics and functions as a “hydride shuttle”. This synergistic interaction provides a system that is electrocatalytic with respect to the carbon-based Lewis acid, decreases the required potential for hydrogen oxidation by 1 V, and can be recycled multiple times.

![BORANE, TRIS[2,4-BIS(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/681900/681812-06-6.png)
![BORANE, TRIS[2,4-BIS(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/681900/681812-06-6_b.png)
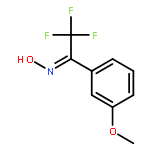
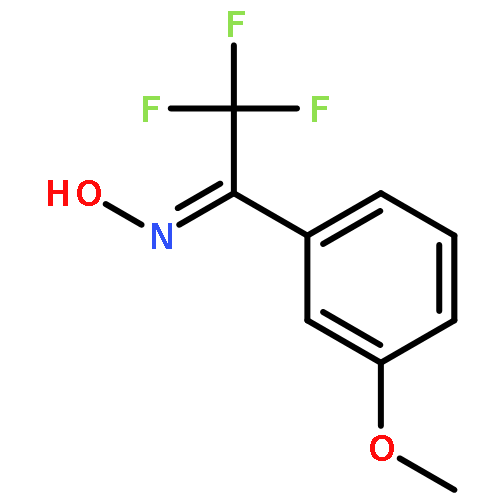
![Phenol, 3-[3-(trifluoromethyl)-3H-diazirin-3-yl]-](http://img.cochemist.com/ccimg/113800/113787-85-2.png)
![Phenol, 3-[3-(trifluoromethyl)-3H-diazirin-3-yl]-](http://img.cochemist.com/ccimg/113800/113787-85-2_b.png)
![Borane, tris[3,5-bis(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/169200/169116-84-1.png)
![Borane, tris[3,5-bis(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/169200/169116-84-1_b.png)