Co-reporter:Soham Dutta
Chemical Society Reviews 2017 vol. 46(Issue 24) pp:7787-7839
Publication Date(Web):2017/12/11
DOI:10.1039/C7CS00555E
Research on surface chirality is motivated by the need to develop functional chiral surfaces for enantiospecific applications. While molecular chirality in 3D has been the subject of study for almost two centuries, many aspects of 2D chiral surface chemistry have yet to be addressed. In 3D, racemic mixtures of chiral molecules tend to aggregate into racemate (molecularly heterochiral) crystals much more frequently than conglomerate (molecularly homochiral) crystals. Whether chiral adsorbates on surfaces preferentially aggregate into heterochiral rather than homochiral domains (2D crystals or clusters) is not known. In this review, we have made the first attempt to answer the following question based on available data: in 2D racemic mixtures adsorbed on surfaces, is there a clear preference for homochiral or heterochiral aggregation? The current hypothesis is that homochiral packing is preferred on surfaces; in contrast to 3D where heterochiral packing is more common. In this review, we present a simple hierarchical scheme to categorize the chirality of adsorbate–surface systems. We then review the body of work using scanning tunneling microscopy predominantly to study aggregation of racemic adsorbates. Our analysis of the existing literature suggests that there is no clear evidence of any preference for either homochiral or heterochiral aggregation at the molecular level by chiral and prochiral adsorbates on surfaces.
Co-reporter:Matthew A. Payne, James B. Miller, Andrew J. Gellman
Corrosion Science 2016 Volume 106() pp:61-81
Publication Date(Web):May 2016
DOI:10.1016/j.corsci.2016.01.026
•AlxFeyNi1 − x − y (x = 0 → 1, y = 0 → 1 − x) oxidation in H2O/air at 427 °C was studied.•The continuous composition boundary of internal/external oxidation was identified.•Data were compared with AlxFeyNi1−x-y oxidation in dry air at 427 °C.•The differences are assessed in terms of a modified Wagner–Maak criterion.The presence of H2O vapour in an oxidizing environment can increase the critical aluminium concentration, NAl*, required to establish a passivating Al2O3 scale in multicomponent alumina-forming alloys. This work used AlxFeyNi1 − x − y composition spread alloy film combinatorial libraries to determine NAl*(x, y) continuously across AlxFeyNi1 − x − y composition space (x = 0 → 1, y = 0 → [1 − x]) in a 10% H2O/air mixture at 427 °C. The NAl*(x, y) in this environment was significantly higher across much of composition space than that previously measured in dry air. Physical insights from the observed differences are considered using a modified Wagner–Maak model.
Co-reporter:Matthew A. Payne, James B. Miller, and Andrew J. Gellman
ACS Combinatorial Science 2016 Volume 18(Issue 9) pp:559
Publication Date(Web):July 5, 2016
DOI:10.1021/acscombsci.6b00047
Composition spread alloy films (CSAFs) are commonly used as libraries for high-throughput screening of composition-property relationships in multicomponent materials science. Because lateral gradients afford two degrees of freedom, an n-component CSAF can, in principle, contain any composition range falling on a continuous two-dimensional surface through an (n – 1)-dimensional composition space. However, depending on the complexity of the CSAF gradients, characterizing and graphically representing this composition range may not be straightforward when n ≥ 4. The standard approach for combinatorial studies performed using quaternary or higher-order CSAFs has been to use fixed stoichiometric ratios of one or more components to force the composition range to fall on some well-defined plane in the composition space. In this work, we explore the synthesis of quaternary Al–Fe–Ni–Cr CSAFs with a rotatable shadow mask CSAF deposition tool, in which none of the component ratios are fixed. On the basis of the unique gradient geometry produced by the tool, we show that the continuous quaternary composition range of the CSAF can be rigorously represented using a set of two-dimensional “pseudoternary” composition diagrams. We then perform a case study of (AlxFeyNi1–x–y)∼0.8Cr∼0.2 oxidation in dry air at 427 °C to demonstrate how such CSAFs can be used to screen an alloy property across a continuous two-dimensional subspace of a quaternary composition space. We identify a continuous boundary through the (AlxFeyNi1–x–y)∼0.8Cr∼0.2 subspace at which the oxygen uptake into the CSAF between 1 and 16 h oxidation time increases abruptly with decreasing Al content. The results are compared to a previous study of the oxidation of AlxFeyNi1–x–y CSAFs in dry air at 427 °C.Keywords: Al−Fe−Ni−Cr; composition gradient films; high-throughput screening; oxidation; quaternary materials libraries
Co-reporter:Matthew A. Payne, James B. Miller, Martin E. Oliveros, Geronimo Perez, Cristol P. Gouvea, Bráulio S. Archanjo, Carlos A. Achete, and Andrew J. Gellman
ACS Combinatorial Science 2016 Volume 18(Issue 7) pp:425
Publication Date(Web):May 25, 2016
DOI:10.1021/acscombsci.6b00030
The high-temperature oxidation of multicomponent metal alloys exhibits complex dependencies on composition, which are not fully understood for many systems. Combinatorial screening of the oxidation of many different compositions of a given alloy offers an ideal means for gaining fundamental insights into such systems. We have previously developed a high-throughput methodology for studying AlxFeyNi1–x–y alloy oxidation using ∼100 nm thick composition spread alloy films (CSAFs). In this work, we critically assess two aspects of this methodology: the sensitivity of CSAF oxidation behavior to variations in AlxFeyNi1–x–y composition and the differences between the oxidation behavior of ∼100 nm thick CSAFs and that of bulk AlxFeyNi1–x–y alloys. This was done by focusing specifically on AlxFe1–x and AlxNi1–x oxidation in dry air at 427 °C. Transitions between phenomenologically distinguishable types of oxidation behavior are found to occur over CSAF compositional ranges of <2 at. %. The oxidation of AlxFe1–x CSAFs is found to be very similar to that of bulk AlxFe1–x alloys, but some minor differences between CSAF and bulk behavior are observed for AlxNi1–x oxidation. On the basis of our assessment, high-throughput studies of CSAF oxidation appear to be an effective method for gaining fundamental insights into the composition dependence of the oxidation of bulk alloys.Keywords: Al2O3 passivation; Al−Fe−Ni; high-throughput screening; materials libraries; oxidation; thin films
Co-reporter:Andrew J. Gellman, L. Baker, B.S. Holsclaw
Surface Science 2016 Volume 646() pp:83-89
Publication Date(Web):April 2016
DOI:10.1016/j.susc.2015.10.062
•Xe adsorption on flat, stepped and kinked Pt single crystal surfaces•Determination of terrace, step and kink site distributions on Pt surfaces• Measurement of areal density of chiral kink sites on Pt(531)RThe ideal structures of the Pt(111), Pt(221) and Pt(531) surfaces expose adsorption sites that can be qualitatively described as terrace sites on Pt(111), both step and terrace sites on Pt(221), and kink sites on Pt(531). The real surface structures of these surfaces can be complicated by imperfections such as misorientation, reconstruction and thermal roughening, all of which will influence their distributions of adsorption sites. Xe adsorption sites on the Pt(111), Pt(221) and Pt(531) surfaces have been probed using both photoemission of adsorbed Xe (PAX) and temperature programmed desorption (TPD) of Xe. Both PAX and Xe TPD are sensitive to the adsorption sites of the Xe and serve as complementary means of assessing the distributions of adsorption sites on these three Pt surfaces. The adsorption of Xe is sufficiently sensitive to detect the presence of residual steps on the Pt(111) surface at a density of ~ 1.5% step atoms per Pt atom. On the Pt(221) surface, PAX and Xe TPD reveal adsorption at both terrace and step sites simultaneously. Although the ideal structure of the Pt(531) surface has no well-defined steps or terraces, Xe adsorption indicates that its adsorption sites are best described as a distribution of both step and kink sites with roughly twice as many steps sites as kinks.
Co-reporter:N. Shukla, D. Yang, A.J. Gellman
Surface Science 2016 Volume 648() pp:29-34
Publication Date(Web):June 2016
DOI:10.1016/j.susc.2015.10.061
•Enantiospecific adsorption of a chiral pharmaceutical on chiral Au nanoparticles•Application of optical polarimetry to detection and analysis of enantioselective adsorption•Quantitative determination of enantiospecific adsorption equilibrium constants.Tetrahexahedral (THH, 24-sided) Au nanoparticles modified with D- or L-cysteine (Cys) have been used as enantioselective separators of the chiral pharmaceutical propranolol (PLL) in solution phase. Polarimetry has been used to measure the rotation of linearly polarized light by solutions containing mixtures of PLL and Cys/THH–Au NPs with varying enantiomeric excesses of each. Polarimetry yields clear evidence of enantiospecific adsorption of PLL onto the Cys/THH–Au NPs. This extends prior work using propylene oxide as a test chiral probe, by using the crystalline THH Au NPs with well-defined facets to separate a real pharmaceutical. This work suggests that chiral nanoparticles, coupled with a density separation method such as centrifugation, could be used for enantiomeric purification of real pharmaceuticals. A simple robust model developed earlier has also been used to extract the enantiospecific equilibrium constants for R- and S-PLL adsorption onto the D- and L-Cys/THH–Au NPs.
Co-reporter:Yongju Yun and Andrew J. Gellman
The Journal of Physical Chemistry C 2016 Volume 120(Issue 48) pp:27285-27295
Publication Date(Web):November 3, 2016
DOI:10.1021/acs.jpcc.6b07758
The enantiospecific adsorption of enantiomer mixtures on surfaces is dictated by two competing forces: the enantiospecificity of adsorption energetics and the propensity of enantiomers to aggregate into homochiral (conglomerate) or heterochiral (racemate) clusters. These phenomena have been studied by measuring the surface enantiomeric excess, ees, of chiral amino acid mixtures adsorbed on Cu single-crystal surfaces and in equilibrium with gas-phase mixtures of varying enantiomeric excess, eeg. Alanine adsorption on Cu{3,1,17}R&S surfaces is nonenantiospecific, ees = eeg, because alanine enantiomers do not interact with either the surface or with one another enantiospecifically. Aspartic acid adsorbs enantiospecifically on the Cu{3,1,17}R&S surfaces; ees ≠ eeg, even during exposure to a racemic mixture in the gas phase, eeg = 0. Exposure of the achiral Cu{111} surface to nonracemic aspartic acid, eeg ≠ 0, results in local amplification of enantiomeric excess on the surface, |ees| > |eeg|, as a result of homochiral aggregation. Finally, despite the fact that the Cu{653}R&S surfaces are chiral, the adsorption of aspartic acid mixtures yields |ees| > |eeg|, indicating that homochiral aggregation dominates enantiospecific adsorbate–surface interactions. All of these types of behavior are captured by a Langmuir-like adsorption isotherm that includes competition between enantiospecific adsorption and both homochiral (conglomerate) and heterochiral (racemate) aggregation of chiral adsorbates.
Co-reporter:G. Gumuslu, P. Kondratyuk, J. R. Boes, B. Morreale, J. B. Miller, J. R. Kitchin, and A. J. Gellman
ACS Catalysis 2015 Volume 5(Issue 5) pp:3137
Publication Date(Web):April 7, 2015
DOI:10.1021/cs501586t
The relationship between alloy catalyst activity and valence band electronic structure has been investigated experimentally across a broad, continuous span of CuxPd1–x composition space. CuxPd1–x composition spread alloy films (CSAFs) were used as catalyst libraries with a 100 channel microreactor to measure the H2–D2 exchange kinetics over a temperature range of 333–593 K at 100 discrete CuxPd1–x compositions spanning the range x = 0.30–0.97. The H2–D2 exchange activity exhibits a monotonic decrease over the composition range x = 0.30–0.97. A steady state, microkinetic model was used to estimate the energy barriers to dissociative H2 adsorption, ΔEads‡, and recombinative H2 desorption, ΔEdes‡, as functions of alloy composition, x. Their values fall in the ranges ΔEads‡(x) = 0.15 to 0.45 eV and ΔEdes‡(x) = 0.55–0.65 eV. Spatially resolved UV photoemission spectra were obtained from the CuxPd1–x CSAF and used to estimate the average energy of the filled states of the valence band as a function of alloy composition, εv(x). The energy of the v-band center shifted monotonically from εv = −3.3 to −3.9 eV across the composition range x = 0.30–0.97. This monotonic shift and its magnitude were corroborated by DFT calculations. The correlation of ΔEads‡(x) with εv(x) across alloy composition space yields ΔEads‡(εv) which decreases as the v-band energy shifts toward the Fermi level.Keywords: adsorption; alloys; catalysis; electronic structure; hydrogen; membranes; palladium
Co-reporter:Yongju Yun and Andrew J. Gellman
Langmuir 2015 Volume 31(Issue 22) pp:6055-6063
Publication Date(Web):May 1, 2015
DOI:10.1021/acs.langmuir.5b00707
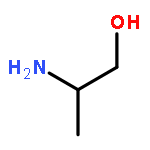
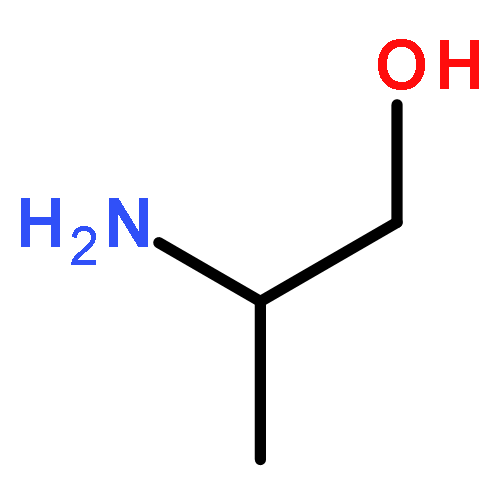
Gas-phase equilibrium adsorption of d- and l-serine (Ser) mixtures and d- and l-phenylalanine (Phe) mixtures has been studied on the naturally chiral Cu{3,1,17}R&S surfaces. 13C labeling of the l enantiomers (*l-Ser and *l-Phe) has enabled mass spectrometric enantiodiscrimination of the species desorbing from the surface following equilibrium adsorption. On the Cu{3,1,17}R&S surfaces, both equilibrium adsorption and the thermal decomposition kinetics of the d and *l enantiomers exhibit diastereomerism. Following exposure of the surfaces to d/*l mixtures, the relative equilibrium coverages of the two enantiomers are equal to their relative partial pressures in the gas phase, θd/θ*l = Pd/P*l. This implies that adsorption is not measurably enantiospecific. The decomposition kinetics of Ser are enantiospecific whereas those of Phe are not. Comparison of these results with those for aspartic acid, alanine, and lysine suggests that enantiospecific adsorption on the naturally chiral Cu surfaces occurs for those amino acids that have side chains with functional groups that allow strong interactions with the surface. There is no apparent correlation between amino acids that exhibit enantiospecific adsorption and those that exhibit enantiospecific decomposition kinetics.
Co-reporter:Aaron Reinicker;James B. Miller;Wooseok Kim;Kijung Yong
Topics in Catalysis 2015 Volume 58( Issue 10-11) pp:613-622
Publication Date(Web):2015 August
DOI:10.1007/s11244-015-0403-z
The decomposition of CH3CH2OH, CD3CD2OD, and CF3CH2OH on Zn\( \left( {1\bar{1}00} \right) \) was studied using temperature programmed reaction spectroscopy. CH3CH=O (CD3CD=O, CF3CH=O), CH2=CH2 (CD2=CD2, CF2=CH2), H2O (D2O) and H2 (D2) were formed in all cases. The CH3CH2OH decomposition mechanism includes the formation of two intermediate species on the surface: CH3CH2- bonded to surface lattice O atoms decomposes to form CH2=CH2 while CH3CH2O– bonded to surface Zn atoms decomposes to form CH3CH=O. A significant isotope effect observed for the formation of CH2=CH2 versus CD2=CD2 suggests that C–H(D) bond breaking at the β-carbon is the rate-limiting step in CH3CH2– (CD3CD2–) decomposition. Decomposition of CF3CH2OH leaves F-atoms on the surface as a result of β-fluoride elimination in CF3CH2–. A significant F substituent effect in desorption of CF3CH=O versus CH3CH=O indicates that the CF3 group increases the barrier to the β-hydride elimination step yielding CF3CH=O and suggests that the transition state is cationic, \( C^{\delta + } \cdots H^{\delta - } \).
Co-reporter:Daniel S. Wei, Bharat S. Mhatre, Andrew J. Gellman, David S. Sholl
Surface Science 2014 Volume 629() pp:35-40
Publication Date(Web):November 2014
DOI:10.1016/j.susc.2014.03.022
•Physisorption on low and high Miller index Cu surfaces was modeled with DFT.•Dispersion corrected DFT shows tendency to overbind the adsorbate.•Calculated results are qualitatively consistent with experimental TPD data.•Calculation indicates possible adsorbate induced surface reconstruction on Cu(110).The physisorption of R-3-methylcyclohexanone on low and high Miller index Cu surfaces is studied with temperature programmed desorption (TPD) and density functional theory (DFT). The DFT calculations are performed with D2, vdW-optB86b, and vdW-optB88 dispersion corrected methods. The adsorption energies calculated by the dispersion corrected methods are more comparable to the TPD results than those calculated without dispersion corrections, although, the former methods have a tendency to overbind the surface adsorbates. The implementation of dispersion corrected methods also indicates a possible adsorbate induced surface reconstruction on Cu(110).
Co-reporter:Nisha Shukla, Nathaniel Ondeck, Andrew J. Gellman
Surface Science 2014 Volume 629() pp:15-19
Publication Date(Web):November 2014
DOI:10.1016/j.susc.2014.03.011
•New method for extraction of enantiospecific adsorption constants from optical rotation.•Enantiospecific separation of propylene oxide on chiral Au nanoparticles.•Measured enantiospecific ratios of adsorption constants for R- and S-PO on D- and L-cys/Au-NP.Au nanoparticles modified with enantiomerically pure d- or l-cysteine have been shown to serve as enantioselective adsorbents of R- and S-propylene oxide. A simple adsorption model and accompanying experimental protocol have been developed to enable optical rotation measurements to be analyzed for quantitative determination of the ratios of the enantiospecific adsorption equilibrium constants of chiral species on the surfaces of chiral nanoparticles, KLS/KDS = KDR/KLR. This analysis is robust in the sense that it obviates the need to measure the absolute surface area of the absorbent nanoparticles, a quantity that is somewhat difficult to obtain accurately. This analysis has been applied to optical rotation data obtained from solutions of R- and S-propylene oxide, in varying concentration ratios, with d- and l-cysteine coated Au nanoparticles, in varying concentration ratios.
Co-reporter:Yongju Yun ; Daniel Wei ; David S. Sholl
The Journal of Physical Chemistry C 2014 Volume 118(Issue 27) pp:14957-14966
Publication Date(Web):June 13, 2014
DOI:10.1021/jp503796u
Equilibrium adsorption of gas phase mixtures of d- and l-alanine (Ala) onto the naturally chiral Cu{3,1,17}R&S surfaces has been studied by both experiment and DFT-based modeling. Isotopically labeled *l-Ala (HO213CCH(NH2)CH3) and unlabeled d-Ala allow mass spectrometric enantiodifferentiation of the adsorbed species during temperature-programmed decomposition, following equilibrium adsorption. Measurements of the relative equilibrium coverages of d- and *l-Ala on the Cu{3,1,17}R&S surfaces, θD/R/θ*L/R = θ*L/S/θD/S, at gas phase partial pressure ratios of P*L/PD = 1/2, 1, and 2 indicate that the d-Ala and *l-Ala conglomerate phases are more energetically stable than a d*l-Ala racemate phase, but that their adsorption energies are not measurably enantiospecific, ΔΔEDL ≈ 0. Although the DFT simulations provide a self-consistent structure of Ala overlayers on Cu{3,1,17}R&S they overestimate the enantiospecificity of the adsorption energetics.
Co-reporter:Andrew J. Gellman ; Ye Huang ; Xu Feng ; Vladimir V. Pushkarev ; Brian Holsclaw ;Bharat S. Mhatre
Journal of the American Chemical Society 2013 Volume 135(Issue 51) pp:19208-19214
Publication Date(Web):November 21, 2013
DOI:10.1021/ja408659v
Chiral inorganic materials predated life on Earth, and their enantiospecific surface chemistry may have played a role in the origins of biomolecular homochirality. However, enantiospecific differences in the interaction energies of chiral molecules with chiral surfaces are small and typically lead to modest enantioselectivities in adsorption, catalysis, and chemistry on chiral surfaces. To yield high enantioselectivities, small energy differences must be amplified by reaction mechanisms such as autocatalytic surface explosions which have nonlinear kinetics. Herein, we report the first observations of superenantiospecificity resulting from an autocatalytic surface explosion reaction of a chiral molecule on a naturally chiral surface. R,R- and S,S-tartaric acid decompose via a vacancy-mediated surface explosion mechanism on Cu single crystal surfaces. When coupled with surface chirality, this leads to decomposition rates that exhibit extraordinarily high enantiospecificity. On the enantiomorphs of naturally chiral Cu(643)R&S, Cu(17,5,1)R&S, Cu(531)R&S and Cu(651)R&S single crystal surfaces, R,R- and S,S-tartaric acid exhibit enantiospecific decomposition rates that differ by as much as 2 orders of magnitude, despite the fact that the effective rates constants for decomposition differ by less than a factor of 2.
Co-reporter:A. de Alwis, B. Holsclaw, V.V. Pushkarev, A. Reinicker, T.J. Lawton, M.E. Blecher, E.C.H. Sykes, A.J. Gellman
Surface Science 2013 Volume 608() pp:80-87
Publication Date(Web):February 2013
DOI:10.1016/j.susc.2012.09.015
A set of six spherically curved Cu single crystals referred to as Surface Structure Spread Single Crystals (S4Cs) has been prepared in such a way that their exposed surfaces collectively span all possible crystallographic surface orientations that can be cleaved from the face centered cubic Cu lattice. The method for preparing these S4Cs and for finding the high symmetry pole point is described. Optical profilometry has been used to determine the true shapes of the S4Cs and show that over the majority of the surface, the shape is extremely close to that of a perfect sphere. The local orientations of the surfaces lie within ± 1° of the orientation expected on the basis of the spherical shape; their orientation is as good as that of many commercially prepared single crystals. STM imaging has been used to characterize the atomic level structure of the Cu(111) ± 11°-S4C. This has shown that the average step densities and the average step orientations match those expected based on the spherical shape. In other words, although there is some distribution of step–step spacing and step orientations, there is no evidence of large scale reconstruction or faceting. The Cu S4Cs have local structures based on the ideal termination of the face centered cubic Cu lattice in the direction of termination. The set of Cu S4Cs will serve as the basis for high throughput investigations of structure sensitive surface chemistry on Cu.Highlights► Preparation of curved surface single crystals with almost perfect sphericity ► First high throughput, STM characterization of surface structures ► Measurement of step densities and orientations on planes vicinal to Cu(111)
Co-reporter:Yongju Yun ; Andrew J. Gellman
Angewandte Chemie 2013 Volume 125( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/ange.201209025
Co-reporter:Yongju Yun ; Andrew J. Gellman
Angewandte Chemie International Edition 2013 Volume 52( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/anie.201209025
Co-reporter:B. S. Mhatre, V. Pushkarev, B. Holsclaw, T. J. Lawton, E. C. H. Sykes, and A. J. Gellman
The Journal of Physical Chemistry C 2013 Volume 117(Issue 15) pp:7577-7588
Publication Date(Web):February 4, 2013
DOI:10.1021/jp3119378
Autocatalytic reaction mechanisms are observed in a range of important chemical processes including catalysis, radical-mediated explosions, and biosynthesis. Because of their complexity, the microscopic details of autocatalytic reaction mechanisms have been difficult to study on surfaces and heterogeneous catalysts. Autocatalytic decomposition reactions of S,S- and R,R-tartaric acid (TA) adsorbed on Cu(110) offer molecular-level insight into aspects of these processes, which until now, were largely a matter of speculation. The decomposition of TA/Cu(110) is initiated by a slow, irreversible process that forms vacancies in the adsorbed TA layer, followed by a vacancy-mediated, explosive decomposition process that yields CO2 and small hydrocarbon products. Initiation of the explosive decomposition of TA/Cu(110) has been studied by measurement of the reaction kinetics, time-resolved low energy electron diffraction (LEED), and time-resolved scanning tunneling microscopy (STM). Initiation results in a decrease in the local coverage of TA and a concomitant increase in the areal vacancy concentration. Observations of explosive TA decomposition on the Cu(651)S surface suggest that initiation does not occur at structural defects in the surface, as has been suggested in the past. Once the vacancy concentration reaches a critical value, the explosive, autocatalytic decomposition step dominates the TA decomposition rate. The onset of the explosive decomposition of TA on Cu(110) is accompanied by the extraction of Cu atoms from the surface to form a (±6,7; ∓2,1) overlayer that is readily observable using LEED and STM. The explosive decomposition step is second-order in vacancy concentration and accelerates with increasing extent of reaction.
Co-reporter:W.D. Michalak, J.B. Miller, D.R. Alfonso, A.J. Gellman
Surface Science 2012 Volume 606(3–4) pp:146-155
Publication Date(Web):February 2012
DOI:10.1016/j.susc.2011.08.022
Understanding the interactions of hydrogen atoms with the surface and the subsurface regions of Pd is critical to the development of advanced energy technologies for hydrogen storage, hydrogen separations, and catalytic conversion processes. While many of the physical and chemical characteristics of the H2–Pd system are known, the kinetics and thermodynamics of H atom absorption into the bulk, transport from the bulk back to the surface, and desorption from the surface remain unclear. In this work, the kinetics of D2 release from Pd following exposure to D2 over a range of pressures and temperatures were measured using temperature programmed desorption. To accurately simulate the kinetics of D2 release, the continuum-based model of Mavrikakis, et al. (J. Chem. Phys 105, 8398, 1996) was extended to include activation barriers for desorption and transport that depend on D atom concentration in the surface, subsurface and bulk regions of the Pd. The use of concentration dependent barriers improves the ability of the model to predict the hydrogen uptake and release kinetics observed across temperatures ranging from 100 to 600 K.Highlights► Development of a transport model to describe H uptake and release from Pd(100). ► Inclusion of concentration dependent transport coefficients and rate constants. ► Prediction of H atom distribution within surface, subsurface and bulk of Pd(100).
Co-reporter:William D. Michalak, James B. Miller, Cem Yolcu, Andrew J. Gellman
Thin Solid Films 2012 Volume 522() pp:473-479
Publication Date(Web):1 November 2012
DOI:10.1016/j.tsf.2012.07.041
Metal nanoparticles on structured supports are used in a variety of technological applications including biosensing, energy harvesting, and electronics. In every case, the functions and properties of the metallic nanostructures depend on both their composition and structure (i.e. size, shape, and spatial distribution). Among the challenges to the development of metal nanoparticles for these applications is the characterization of relationships between their structure and their functional properties over multiple structural degrees of freedom spanning a large range of values. In this work, a method for creating a morphological gradient of metal nanoparticles on a substrate is described. The approach, suited for high-throughput fabrication and characterization, is based on spinodal dewetting of a metallic thin film from its substrate. Through control of initial film thickness, anneal temperature, and anneal time, spinodal dewetting results in supported nanoparticles with well-defined and controlled structure. The approach is demonstrated through its application to preparation of Pd nanoparticles on a silicon nitride substrate. The morphologies of the particles were characterized by scanning electron and atomic force microscopies. Free energy-based stability and topological analyses were used to confirm the dewetting mechanism. In addition, the stability theory provides a connection to the thermophysical properties of the resulting nanoparticle array. The dewetting approach is general to any metal/support system and provides an alternative, inexpensive, and robust means to rapidly create metal nanostructures with control of morphology. It shows promise for large scale production of metal nanoparticles structures, as well as understanding basic stability properties of thin metal films.Highlights► Pd dewetting from SiN occurs by a spinodal dewetting mechanism. ► Dewetting occurs at temperatures well below the melting point of Pd. ► Spinodal dewetting allows predictable control of metal particle size and morphology. ► High throughput experimental method.
Co-reporter:Wai Yeng Cheong and Andrew J. Gellman
Langmuir 2012 Volume 28(Issue 43) pp:15251-15262
Publication Date(Web):September 28, 2012
DOI:10.1021/la3027557
The enantiospecific desorption kinetics of R- and S-propylene oxide (PO) from a Cu(100) surface modified by enantiomerically pure d- or l-lysine have been studied using temperature programmed desorption. These experiments have used R- or S-PO as the chiral probe for study of enantiospecific adsorption on Cu(100) surfaces modified with d- or l-lysine. This chiral probe/modifier/Cu system manifests a significant diastereomeric effect in the R- and S-PO peak desorption temperatures and, hence, true enantiospecific behavior. The enantiospecificity in the PO desorption kinetics is observed only over a narrow range of lysine modifier coverage with a maximum at a lysine coverage leaving an empty site density of θO ≈ 0.25. The observation of enantiospecific behavior in the PO/lysine/Cu(100) system is in contrast with the failed results of prior attempts to observe enantiospecific desorption from chirally modified Cu surfaces. The potential for hydrogen-bonding interactions between the chiral probe and chiral modifier, which can depend on the coverage and configuration of the adsorbed modifier, may play a crucial role in enantiospecific adsorption on lysine modified Cu surfaces.
Co-reporter:T.J. Lawton, V. Pushkarev, E. Broitman, A. Reinicker, E.C.H. Sykes, and A.J. Gellman
The Journal of Physical Chemistry C 2012 Volume 116(Issue 30) pp:16054-16062
Publication Date(Web):July 6, 2012
DOI:10.1021/jp303488t
The initial stage in the oxidation of Cu single crystal surfaces has been studied on a surface structure spread single crystal (S4C) exposing a continuous distribution of all Cu(hkl) surface orientations lying within 10° polar angle of the (111) plane, Cu(111) ± 10°-S4C. The uptake of oxygen across the Cu(111) ± 10°-S4C during exposure to O2 at 300 K has been measured using spatially resolved X-ray photoelectron spectroscopy (XPS), and the resulting Cu2O surface oxide layer has been imaged using scanning tunneling microscopy (STM). Uptake of oxygen is dependent on surface step density and increases with increasing polar angle relative to the (111) pole. In contrast, the oxygen uptake does not depend on the crystallographic orientation of the step edge or, in other words, the kink density along the step edge. STM images reveal that once oxidation of the step edges begins, all of the boundaries of the Cu2O step oxide layer are oriented along (100) step edges in the Cu(111) terrace independent of the initial orientation of the step. In other words, the oxidizing step edges have no memory of their original orientation, and thus, the step growth depends only on step density and not on the kink density along the step edge. The combined use of both spatially resolved XPS and atomic scale imaging with STM on a Cu(111) ± 10°-S4C has provided unique insight into the origins of structure-sensitive surface chemistry.
Co-reporter:Wai Yeng Cheong
The Journal of Physical Chemistry C 2011 Volume 115(Issue 4) pp:1031-1035
Publication Date(Web):September 8, 2010
DOI:10.1021/jp105520t
Temperature-programmed desorption experiments have been used to probe the adsorption energetics of d- and l-lysine on the chiral Cu(3,1,17)R&S and achiral Cu(100) surfaces. Previous literature has reported the reconstruction of Cu(100) surfaces to form homochiral (3,1,17)R facets upon adsorption of l-lysine at high coverage and after annealing to 430 K (J. Am. Chem. Soc. 122, 2000, 12584). The implication of that work is that the adsorption energy of l-lysine on Cu(3,1,17)R is greater than on Cu(3,1,17)S and Cu(100), thereby driving the homochiral reconstruction. The results of temperature-programmed desorption measurements test and support this implication. The desorption energies of d- and l-lysine on the Cu(100) surface are significantly lower than those on either of the Cu(3,1,17)R&S surfaces. Furthermore, large enantiospecific differences in desorption kinetics were observed for d- and l-lysine on the Cu(3,1,17)R&S surfaces. This observed enantiospecificity is believed to originate from the enantiospecific interactions of lysine with the chiral kinked steps on these surfaces. The observation that adsorption of l-lysine on Cu(3,1,17)R is energetically preferred over adsorption on Cu(3,1,17)S is consistent with the formation of homochiral (3,1,17)R facets during l-lysine adsorption on Cu(100).
Co-reporter:Ye Huang
Topics in Catalysis 2011 Volume 54( Issue 19-20) pp:1403-1413
Publication Date(Web):2011/12/01
DOI:10.1007/s11244-011-9756-0
(R)-3-methylcyclohexanone (R-3MCHO) has been shown to adsorb enantiospecifically on naturally chiral Cu surfaces vicinal to the Cu(110) plane. Adsorption of R-3MCHO on seven Cu single crystal surfaces vicinal to (110) was studied using temperature programmed desorption. These surfaces include Cu(110), Cu(771), Cu(430), Cu(13,9,1)R&S and Cu(651)R&S. The Cu(13,9,1)R&S and Cu(651)R&S surfaces are naturally chiral surfaces with terrace-step-kink structures. Enantioselective adsorption of R-3MCHO takes place on the chiral kink sites of these surfaces. Three R-3MCHO desorption features were resolved in the TPD spectra on Cu(13,9,1)R&S and Cu(651)R&S surfaces. Based upon comparisons between these and other Cu single crystal surfaces, they were assigned to desorption of R-3MCHO from flat terrace, close-packed step and kink sites. The desorption of R-3MCHO from the row and trough structure of the Cu(110) surface resembled desorption from a step structure rather than from a flat Cu(111) terrace. R-3MCHO desorbs enantiospecifically from the Cu(13,9,1)R&S and Cu(651)R&S surfaces. The peaks associated with R-3MCHO desorbing from the R- and S-chiral kink sites on Cu(13,9,1)R&S differed in temperature by 2.4 ± 0.8 K. This corresponds to an enantiospecific difference in the desorption energies of 0.7 ± 0.2 kJ/mol, with a preference for R-3MCHO adsorption at the R-kinks. In contrast, R-3MCHO has a desorption energy from the S-kinks on the Cu(651)S surface that is 0.7 ± 0.2 kJ/mol higher than from the R-kinks on the Cu(651)R surface.
Co-reporter:Nisha Shukla ; Melissa A. Bartel
Journal of the American Chemical Society 2010 Volume 132(Issue 25) pp:8575-8580
Publication Date(Web):June 3, 2010
DOI:10.1021/ja908219h
The surfaces of chemically synthesized Au nanoparticles have been modified with d- or l-cysteine to render them chiral and enantioselective for adsorption of chiral molecules. Their enantioselective interaction with chiral compounds has been probed by optical rotation measurements during exposure to enantiomerically pure and racemic propylene oxide. The ability of optical rotation to detect enantiospecific adsorption arises from the fact that the specific rotation of polarized light by (R)- and (S)-propylene oxide is enhanced by interaction with Au nanoparticles. This effect is related to previous observations of enhanced circular dichroism by Au nanoparticles modified by chiral adsorbates. More importantly, chiral Au nanoparticles modified with either d- or l-cysteine selectively adsorb one enantiomer of propylene oxide from a solution of racemic propylene oxide, thus leaving an enantiomeric excess in the solution phase. Au nanoparticles modified with l-cysteine (d-cysteine) selectively adsorb the (R)-propylene oxide ((S)-propylene oxide). A simple model has been developed that allows extraction of the enantiospecific equilibrium constants for (R)- and (S)-propylene oxide adsorption on the chiral Au nanoparticles.
Co-reporter:Andrew J. Gellman
ACS Nano 2010 Volume 4(Issue 1) pp:5
Publication Date(Web):January 26, 2010
DOI:10.1021/nn901885n
Chiral surfaces serve as media for enantioselective chemical processes. Their chirality is dictated by atomic- and molecular-level structure, and their enantioselectivity is determined by their enantiospecific interactions with chiral adsorbates. This Perspective describes three types of chiral metal surfaces: those modified by adsorption of chiral molecules, those templated by chiral lattices of adsorbed species, and those that are naturally chiral. A new paper in this issue of ACS Nano offers insight into the intermolecular interactions that govern chiral templating of surfaces. This Perspective then outlines three major challenges to the field of chiral surface science: development of methods for detection of enantiospecific interactions and enantioselective surface chemistry, preparation of high-area chiral metal surfaces, and the development of a fundamental, predictive-level understanding of the origin of enantioselectivity on chiral surfaces.
Co-reporter:Layton Baker ; Brian Holsclaw ; Ashleigh E. Baber ; Heather L. Tierney ; E. Charles H. Sykes
The Journal of Physical Chemistry C 2010 Volume 114(Issue 43) pp:18566-18575
Publication Date(Web):October 12, 2010
DOI:10.1021/jp106489f
Xe has been used to probe the distributions of adsorption sites across three different Cu single-crystal surfaces: Cu(111), Cu(221), and Cu(643). These expose terrace, step, and kink sites, respectively. The study couples the use of scanning tunneling microscopy (STM), temperature-programmed desorption (TPD), and photoemission of adsorbed Xe (PAX) to assess their use as methods for determining adsorption site distributions on Cu surfaces. STM shows that the Xe adsorption sites in order of energetic preference are kink, step edge, and terrace, but indicates that the binding energy differences between the three are likely very small. This is borne out by Xe TPD studies that show distinct differences in the desorption kinetics on the three surfaces but unresolvable differences in the desorption temperatures and binding energies at the terrace, step, and kink sites. PAX spectra reveal observable features that can be associated with Xe adsorption at terrace, step, and kink sites. These features can be analyzed semiquantitatively to give insight into the distributions of sites on these surfaces.
Co-reporter:Wai Yeng Cheong, Ye Huang, Nikunj Dangaria, and Andrew J. Gellman
Langmuir 2010 Volume 26(Issue 21) pp:16412-16423
Publication Date(Web):July 15, 2010
DOI:10.1021/la102074a
Temperature programmed desorption methods have been used to probe the enantioselectivity of achiral Cu(100), Cu(110), and Cu(111) single crystal surfaces modified by chiral organic molecules including amino acids, alcohols, alkoxides, and amino-alcohols. The following combinations of chiral probes and chiral modifiers on Cu surfaces were included in this study: propylene oxide (PO) on l-alanine modified Cu(110), PO on l-alaninol modified Cu(111), PO on 2-butanol modified Cu(111), PO on 2-butoxide modified Cu(100), PO on 2-butoxide modified Cu(111), R-3-methylcyclohexanone (R-3-MCHO) on 2-butoxide modified Cu(100), and R-3-MCHO on 2-butoxide modified Cu(111). In contrast with the fact that these and other chiral probe/modifier systems have exhibited enantioselectivity on Pd(111) and Pt(111) surfaces, none of these probe/modifier/Cu systems exhibit enantioselectivity at either low or high modifier coverages. The nature of the underlying substrate plays a significant role in the mechanism of hydrogen-bonding interactions and could be critical to observing enantioselectivity. While hydrogen-bonding interactions between modifier and probe molecule are believed to induce enantioselectivity on Pd surfaces (Gao, F.; Wang, Y.; Burkholder, L.; Tysoe, W. T. J. Am. Chem. Soc. 2007, 129, 15240−15249), such critical interactions may be missing on Cu surfaces where hydrogen-bonding interactions are believed to occur between adjacent modifier molecules, enabling them to form clusters or islands.
Co-reporter:Yang Yun, Esteban Broitman and Andrew J. Gellman
Langmuir 2010 Volume 26(Issue 2) pp:908-914
Publication Date(Web):September 16, 2009
DOI:10.1021/la902375f
Amorphous fluorinated carbon (a-CFx) films have a variety of potential technological applications. In most such applications these films are exposed to air and undergo partial surface oxidation. X-ray photoemission spectroscopy has been used to study the oxidation of fresh a-CFx films deposited by magnetron sputtering. The oxygen sticking coefficient measured by exposure to low pressures (<10−3 Torr) of oxygen at room temperature is on the order of S ≈ 10−6, indicating that the surfaces of these films are relatively inert to oxidation when compared with most metals. The X-ray photoemission spectra indicate that the initial stages of oxygen exposure (<107 langmuirs) result in the preferential oxidation of the carbon atoms with zero or one fluorine atom, perhaps because these carbon atoms are more likely to be found in configurations with unsaturated double bonds and radicals than carbon atoms with two or three fluorine atoms. Exposure of the a-CFx film to atmospheric pressures of air (effective exposure of 1012 langmuirs to O2) results in lower levels of oxygen uptake than the low pressure exposures (<107 langmuirs). It is suggested that this is the result of oxidative etching of the most reactive carbon atoms, leaving a relatively inert surface. Finally, low pressure exposures to air result in the adsorption of both nitrogen and oxygen onto the surface. Some of the nitrogen adsorbed on the surface at low pressures is in a reversibly adsorbed state in the sense that subsequent exposure to low pressures of O2 results in the displacement of nitrogen by oxygen. Similarly, when an a-CFx film oxidized in pure O2 is exposed to low pressures of air, some of the adsorbed oxygen is displaced by nitrogen. It is suggested that these forms of nitrogen and oxygen are bound to free radical sites in the film.
Co-reporter:Joshua D. Horvath ; Layton Baker
The Journal of Physical Chemistry C 2008 Volume 112(Issue 20) pp:7637-7643
Publication Date(Web):April 30, 2008
DOI:10.1021/jp0753878
The high Miller index planes of metal single crystals are chiral, if they do not lie perpendicular to any of the mirror symmetry planes of the bulk lattice. Such chiral surfaces of face-centered cubic metals expose kinked step edges and have been shown to have enantiospecific interactions with chiral adsorbates. R–3-methylcyclohexanone (R-3MCHO) exhibits enantiospecific differences in its desorption energies from the R and S chiral kinks on the Cu(643)R/S surfaces. This enantiospecific interaction must also manifest itself in the orientations of R-3MCHO adsorbed at chiral kinks and has been probed by examining the intensities of infrared absorption by R-3MCHO adsorbed at the kinks on the Cu(643)R/S surfaces. Fourier transform infrared reflection−absorption spectra show that the interaction of the R-3MCHO occurs through the carbonyl group which exhibits a red-shift in its stretching mode as a result of adsorption on the surface. The absorption intensities also indicate that the molecule is oriented with the >C═O bond roughly parallel to the surface. More importantly, R-3MCHO adsorbed at the R and the S kinks on the Cu(643)R/S surfaces exhibits different relative absorption intensities of its vibrational modes, clearly indicating that the orientations of R-3MCHO are enantiospecific on the two enantiomorphic surfaces.
Co-reporter:Ye Huang
Catalysis Letters 2008 Volume 125( Issue 3-4) pp:177-182
Publication Date(Web):2008 October
DOI:10.1007/s10562-008-9600-8
The enantiospecific adsorption and desorption of (R)-3-methylcyclohexanone on naturally chiral Cu(531)R&S surfaces was studied using temperature programmed desorption. The Cu(531)R&S surfaces are of interest because they lie at the center of the stereographic triangle and thus, have the highest density of chiral adsorption sites possible on the surface of a face centered cubic metal. Several (R)-3-methylcyclohexanone desorption features were resolved in the TPD spectra from Cu(531)R&S surfaces and were assigned to desorption of molecules from terrace, step, and kink sites. The peaks associated with (R)-3-methylcyclohexanone desorbing from the R- and S-kink sites differed in temperature by 2.2 ± 0.6 K. This corresponds to an enantiospecific difference in the desorption energies of 0.5 ± 0.2 kJ/mol, with a preference for adsorption of (R)-3-methylcyclohexanone at the S-kinks on the Cu(531)S surface.
Co-reporter:Min Soo Lim, Andrew J. Gellman
Tribology International 2005 Volume 38(6–7) pp:554-561
Publication Date(Web):June–July 2005
DOI:10.1016/j.triboint.2005.01.006
Heat assisted magnetic recording (HAMR) on magnetic hard disks is being explored as a means of increasing the areal density of stored data beyond the limits of current technologies. HAMR will subject the magnetic media, the overcoat, and the lubricant on its surface to temperatures in the range 400–650 °C for periods of a few nanoseconds per pass of the read-write head. During such rapid heating events the lubricant is prone to decomposition and desorption from the surface, either of which lead to degradation of the lubricant film, jeopardizing the integrity of the stored data. Rapid laser annealing is known to bias the reactions of small molecules adsorbed on surfaces to favor desorption over decomposition. Analysis of the desorption and decomposition kinetics of perfluoropolyalkylether lubricants such as Fomblin Zdol shows that rapid heating to high temperatures favors desorption over decomposition for molecules with molecular weights of less than 3000. For higher molecular weight Fomblins decomposition is favored at the temperatures to be used for HAMR.
Co-reporter:Petro Kondratyuk, Gamze Gumuslu, Shantanu Shukla, James B. Miller, Bryan D. Morreale, Andrew J. Gellman
Journal of Catalysis (April 2013) Volume 300() pp:55-62
Publication Date(Web):1 April 2013
DOI:10.1016/j.jcat.2012.12.015
We describe a 100-channel microreactor array capable of spatially resolved measurement of catalytic activity across the surface of a flat substrate. When used in conjunction with a composition spread alloy film (CSAF, e.g., PdxCuyAu1−x−y) across which component concentrations vary smoothly, such measurements permit high-throughput analysis of catalytic activity and selectivity as a function of catalyst composition. In the reported implementation, the system achieves spatial resolution of 1 mm2 over a 10 × 10 mm2 area. During operation, the reactant gases are delivered at constant flow rate to 100 points of differing composition on the CSAF surface by means of a 100-channel microfluidic device. After coming into contact with the CSAF catalyst surface, the product gas mixture from each of the 100 points is withdrawn separately through a set of 100 isolated channels for analysis using a mass spectrometer. We demonstrate the operation of the device on a PdxCuyAu1−x−y CSAF catalyzing the H2–D2 exchange reaction at 333 K. In essentially a single experiment, we measured the catalytic activity over a broad swathe of concentrations from the ternary composition space of the PdxCuyAu1−x−y alloy.Graphical abstractWe describe a glass microreactor array for parallel measurement of catalytic activity at 100 points of different composition across a composition spread alloy film of 1 cm2 area. Study of H2 + D2 → 2HD reveals the continuous variation of catalytic activity across PdxCuyAu1−x−y composition space.Download high-res image (110KB)Download full-size imageHighlights► New 100-channel microreactor array for spatially resolved catalysis across 1 cm2 area. ► Preparation of a PdxCuyAu1−x−y composition spread alloy catalyst spanning composition space. ► H2 + D2 → 2HD on PdxCuyAu1−x−y reveals continuous variation in activity with composition.
Co-reporter:A. D. Reinicker, A. J. Therrien, T. J. Lawton, R. Ali, E. C. H. Sykes and A. J. Gellman
Chemical Communications 2016 - vol. 52(Issue 75) pp:NaN11266-11266
Publication Date(Web):2016/08/18
DOI:10.1039/C6CC05957K
On surfaces vicinal to Cu{111}, L-aspartic acid (L-Asp) adsorption causes steps to facet enantiospecifically into {310}R and {320}S steps. L-Asp has its highest heat of adsortion on surfaces that naturally expose the {310}R or {320}S steps but decomposes preferentially on the {310}R steps.
Co-reporter:B. S. Mhatre, S. Dutta, A. Reinicker, B. Karagoz and A. J. Gellman
Chemical Communications 2016 - vol. 52(Issue 98) pp:NaN14128-14128
Publication Date(Web):2016/11/08
DOI:10.1039/C6CC06887A
Aspartic acid adsorbed on Cu surfaces is doubly deprotonated. On chiral Cu(643)R&S its enantiomers undergo enantiospecific decomposition via an autocatalytic explosion. Once initiated, the decomposition mechanism proceeds via sequential cleavage of the C3–C4 and C1–C2 bonds each yielding CO2, followed by conversion of the remaining species into NCCH3.