Co-reporter:Heming Jiang, Tian-Yu Sun, Xiao Wang, Yaoming Xie, Xinhao Zhang, Yun-Dong Wu, and Henry F. Schaefer III
Organic Letters December 15, 2017 Volume 19(Issue 24) pp:6502-6502
Publication Date(Web):November 22, 2017
DOI:10.1021/acs.orglett.7b03167
2-Iodoxybenzoic acid (IBX) is an important species for the oxidation of alcohols to aldehydes or ketones. An often-cited mechanism involving a hypervalent twist as the rate-determining step (RDS) is inconsistent with kinetic isotope effect (KIE) experiments. The computations with larger basis sets reveal that the reductive elimination involving the C–H bond cleavage is the RDS (rate-determining step). Further computational/experimental studies suggest that the reactivity can be improved by adjusting the trans influence with Lewis acids.
Co-reporter:Juan ZengFan Jiang, Yun-Dong Wu
Journal of Chemical Theory and Computation 2017 Volume 13(Issue 1) pp:
Publication Date(Web):November 17, 2016
DOI:10.1021/acs.jctc.6b00848
Site-specific phosphorylation of an intrinsically disordered protein, eIF4E-binding protein isoform 2 (4E-BP2), can suppress its native function by folding it into a four-stranded β-sheet, but the mechanism of this phosphorylation-induced folding is unclear. In this work, we use all-atom molecular dynamics simulations to investigate both the folded and unfolded states of 4E-BP2 under different phosphorylation states of T37 and T46. The results show that the phosphorylated forms of both T37 and T46 play important roles in stabilizing the folded structure, especially for the β-turns and the sequestered binding motif. The phosphorylated residues not only guide the folding of the protein through several intermediate states but also affect the conformational distribution of the unfolded ensemble. Significantly, the phosphorylated residues can function as nucleation sites for the folding of the protein by forming certain local structures that are stabilized by hydrogen bonding involving the phosphate group. The region around phosphorylated T46 appears to fold before that around phosphorylated T37. These findings provide new insight into the intricate effects of protein phosphorylation.
Co-reporter:Xinhao Zhang, Lung Wa Chung, and Yun-Dong Wu
Accounts of Chemical Research 2016 Volume 49(Issue 6) pp:1302
Publication Date(Web):June 7, 2016
DOI:10.1021/acs.accounts.6b00093
With new advances in theoretical methods and increased computational power, applications of computational chemistry are becoming practical and routine in many fields of chemistry. In organic chemistry, computational chemistry plays an indispensable role in elucidating reaction mechanisms and the origins of various selectivities, such as chemo-, regio-, and stereoselectivities. Consequently, mechanistic understanding improves synthesis and assists in the rational design of new catalysts. In this Account, we present some of our recent works to illustrate how computational chemistry provides new mechanistic insights for improvement of the selectivities of several organic reactions. These examples include not only explanations for the existing experimental observations, but also predictions which were subsequently verified experimentally.This Account consists of three sections discuss three different kinds of selectivities. The first section discusses the regio- and stereoselectivities of hydrosilylations of alkynes, mainly catalyzed by [Cp*Ru(MeCN)3]+ or [CpRu(MeCN)3]+. Calculations suggest a new mechanism that involves a key ruthenacyclopropene intermediate. This mechanism not only explains the unusual Markovnikov regio-selectivity and anti-addition stereoselectivity observed by Trost and co-workers, but also motivated further experimental investigations. New intriguing experimental observations and further theoretical studies led to an extension of the reaction mechanism. The second section includes three cases of meta-selective C–H activation of aryl compounds. In the case of Cu-catalyzed selective meta-C–H activation of aniline, a new mechanism that involves a Cu(III)-Ar-mediated Heck-like transition state, in which the Ar group acts as an electrophile, was proposed. This mechanism predicted a higher reactivity for more electron-deficient Ar groups, which was supported by experiments. For two template-mediated, meta-selective C–H bond activations catalyzed by Pd(II), different mechanisms were derived for the two templates. One involves a dimeric Pd–Pd or Pd–Ag active catalyst, and the other involves a monomeric Pd catalyst, in which a monoprotected amino acid coordinates in a bidentate fashion and serves as an internal base for C–H activation. The third section discusses a desymmetry strategy in asymmetric synthesis. The construction of rigid skeletons is critical for these catalysts to distinguish two prochiral groups. Overall, fruitful collaborations between computational and experimental chemists have provided new and comprehensive mechanistic understanding and insights into these useful reactions.
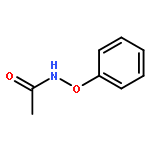
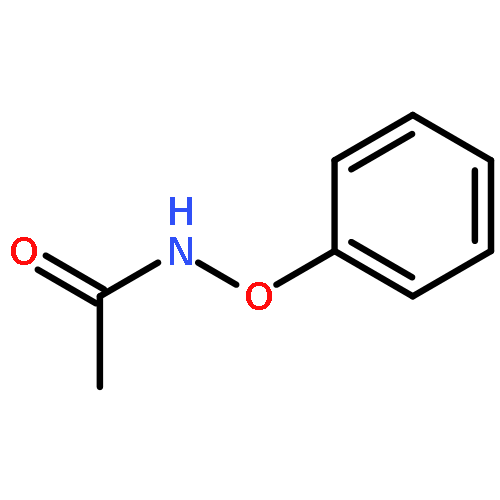
Co-reporter:Yun-Fang Yang; K. N. Houk
Journal of the American Chemical Society 2016 Volume 138(Issue 21) pp:6861-6868
Publication Date(Web):May 13, 2016
DOI:10.1021/jacs.6b03424
The selective rhodium-catalyzed functionalization of arenes is greatly facilitated by oxidizing directing groups that act both as directing groups and internal oxidants. We report density functional theory (B3LYP and M06) investigations on the mechanism of rhodium(III)-catalyzed redox coupling reaction of N-phenoxyacetamides with alkynes. The results elucidated the role of the internal oxidizing directing group, and the role of RhIII/RhI and RhIII/RhV catalysis of C–H functionalizations. A novel RhIII–RhV–RhIII cycle successfully rationalizes recent experimental observations by Liu and Lu et al. (Liu, G. Angew. Chem. Int. Ed. 2013, 52, 6033) on the reactions of N-phenoxyacetamides with alkynes in different solvents. Natural Bond Orbital (NBO) analysis confirms the identity of RhV intermediate in the catalytic cycle.
Co-reporter:Xiu-Mei Zhong, Gui-Juan Cheng, Ping Chen, Xinhao Zhang, and Yun-Dong Wu
Organic Letters 2016 Volume 18(Issue 20) pp:5240-5243
Publication Date(Web):October 4, 2016
DOI:10.1021/acs.orglett.6b02542
A combined mass spectrometric and computational study of the Pd/mono-N-protected amino acid (MPAA)-catalyzed vinyl–vinyl coupling reactions is reported. Computational study reveals that the reaction is initiated by C–H activation of the styrene followed by the insertion of acrylate. This is supported by mass spectrometry. The MPAA ligand facilitates the cross-coupling reaction between monosubstituted alkenes by stabilizing the active palladium catalyst and offering the N-protecting group as a stronger base than acetate. The E/Z selectivity is attributed to the stronger d−π interaction between the catalyst and the substrate in the transition state leading to E product.
Co-reporter:Huan Sun;Yi Zhang;Ping Chen;Xinhao Zhang;Yong Huang
Advanced Synthesis & Catalysis 2016 Volume 358( Issue 12) pp:1946-1957
Publication Date(Web):
DOI:10.1002/adsc.201600015
Co-reporter:Huan Sun;Yi Zhang;Ping Chen;Xinhao Zhang;Yong Huang
Advanced Synthesis & Catalysis 2016 Volume 358( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/adsc.201600460
Co-reporter:Sangni Xun, Fan Jiang, Yun-Dong Wu
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 20) pp:4970-4977
Publication Date(Web):15 October 2016
DOI:10.1016/j.bmc.2016.08.012
RfaH protein functions as both transcription anti-terminator and translation enhancer in bacteria. Recent studies have shown that the C-terminal domain (CTD) is an α-helical hairpin (two-helix bundle) in full-length RfaH, despite the intrinsically favored β-barrel structure. Here, we carried out μs-timescale molecular dynamics (MD) simulations for the wild-type (WT) RfaH, its E48S mutant and an established model without the intrinsically disordered region (IDR1) linking the CTD and the N-terminal domain (NTD). Our simulations showed that the WT can be well stabilized by our RSFF1 force field, while the E48S mutant and the model without IDR1 undergo considerable structural change, which is in good agreement with experimental observations. The IDR1 plays important roles in stabilizing the hydrophobic environment near the crucial E48–R138 salt-bridge as well as in tethering α4 helix in CTD to α3 helix in NTD. In the absence of the IDR1, destabilization of key interdomain contacts and unfolding of the CTD α5 helix were observed in the simulation. In addition, the intrinsically disordered tail of the CTD (IDR2) is also of great significance to stabilize the bound conformation of CTD. These findings provide important implications for consideration of simulations in revealing the functions of residues invisible in a crystal structure.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Le Liu, Tonghuan Zhang, Yun-Fang Yang, Daisy Zhang-Negrerie, Xinhao Zhang, Yunfei Du, Yun-Dong Wu, and Kang Zhao
The Journal of Organic Chemistry 2016 Volume 81(Issue 10) pp:4058-4065
Publication Date(Web):April 28, 2016
DOI:10.1021/acs.joc.6b00345
Combined experimental and theoretical investigations into the phenyliodine bis(trifluoroacetate) (PIFA)-mediated reaction of N-arylcinnamamide to produce 3-arylquinolin-2-one derivatives have been conducted. High regioselectivity during the aryl migration process was observed in 3,3-disubstituted acrylamides. Density functional theory calculation was conducted in an attempt to understand the mechanism and the origin of the regioselectivity. On the basis of both the experimental and the theoretical results, a mechanism involving an oxidative annulation, followed by an aryl migration, has been proposed. The annulation is the regioselectivity determining step.
Co-reporter:Hao Geng; Fan Jiang
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 10) pp:1805-1810
Publication Date(Web):April 29, 2016
DOI:10.1021/acs.jpclett.6b00452
Cyclic peptides (CPs) are promising candidates for drugs, chemical biology tools, and self-assembling nanomaterials. However, the development of reliable and accurate computational methods for their structure prediction has been challenging. Here, 20 all-trans CPs of 5–12 residues selected from Cambridge Structure Database have been simulated using replica-exchange molecular dynamics with four different force fields. Our recently developed residue-specific force fields RSFF1 and RSFF2 can correctly identify the crystal-like conformations of more than half CPs as the most populated conformation. The RSFF2 performs the best, which consistently predicts the crystal structures of 17 out of 20 CPs with rmsd < 1.1 Å. We also compared the backbone (ϕ, ψ) sampling of residues in CPs with those in short linear peptides and in globular proteins. In general, unlike linear peptides, CPs have local conformational free energies and entropies quite similar to globular proteins.
Co-reporter:Li-Juan Song, Shengtao Ding, Yong Wang, Xinhao Zhang, Yun-Dong Wu, and Jianwei Sun
The Journal of Organic Chemistry 2016 Volume 81(Issue 15) pp:6157-6164
Publication Date(Web):May 27, 2016
DOI:10.1021/acs.joc.6b00854
Iridium complexes are known catalysts for a range of silylation reactions. However, the exploitation for selective hydrosilylation of unsymmetrical internal alkynes has been limitedly known. Described here is a new example of this type. Specifically, [(cod)IrCl]2 catalyzes the efficient and mild hydrosilylation of thioalkynes by various silanes with excellent regio- and stereoselectivity. DFT studies suggested a new mechanism involving Ir(I) hydride as the key intermediate.
Co-reporter:Juan Zeng, Fan Jiang, and Yun-Dong Wu
The Journal of Physical Chemistry B 2016 Volume 120(Issue 1) pp:33-41
Publication Date(Web):December 16, 2015
DOI:10.1021/acs.jpcb.5b09027
α-Helical hairpin (two-helix bundle) is a structure motif composed of two interacting helices connected by a turn or a short loop. It is an important model for protein folding studies, filling the gap between isolated α-helix and larger all-α domains. Here, we present, for the first time, successful folding simulations of an α-helical hairpin. Our RSFF1 and RSFF2 force fields give very similar predicted structures of this αtα peptide, which is in good agreement with its NMR structure. Our simulations also give site-specific stability of α-helix formation in good agreement with amide hydrogen exchange experiments. Combining the folding free energy landscapes and analyses of structures sampled in five different ranges of the fraction of native contacts (Q), a folding mechanism of αtα is proposed. The most stable sites of Q9-E15 in helix-1 and E24-A30 in helix-2 close to the loop region act as the folding initiation sites. The formation of interhelix side-chain contacts also initiates near the loop region, but some residues in the central parts of the two helices also form contacts quite early. The two termini fold at a final stage, and the loop region remains flexible during the whole folding process. This mechanism is similar to the “zipping out” pathway of β-hairpin folding.
Co-reporter:Gui-Juan Cheng; Xinhao Zhang; Lung Wa Chung; Liping Xu
Journal of the American Chemical Society 2015 Volume 137(Issue 5) pp:1706-1725
Publication Date(Web):January 8, 2015
DOI:10.1021/ja5112749
Understanding the mechanisms of chemical reactions, especially catalysis, has been an important and active area of computational organic chemistry, and close collaborations between experimentalists and theorists represent a growing trend. This Perspective provides examples of such productive collaborations. The understanding of various reaction mechanisms and the insight gained from these studies are emphasized. The applications of various experimental techniques in elucidation of reaction details as well as the development of various computational techniques to meet the demand of emerging synthetic methods, e.g., C–H activation, organocatalysis, and single electron transfer, are presented along with some conventional developments of mechanistic aspects. Examples of applications are selected to demonstrate the advantages and limitations of these techniques. Some challenges in the mechanistic studies and predictions of reactions are also analyzed.
Co-reporter:Jialing Shi, Ting Wang, Yusha Huang, Xinhao Zhang, Yun-Dong Wu, and Qian Cai
Organic Letters 2015 Volume 17(Issue 4) pp:840-843
Publication Date(Web):February 6, 2015
DOI:10.1021/ol5036613
Employing a chiral spirodiphosphine monoxide ligand with 1,1′-spirobiindane backbone (SDP(O)), a desymmetrization strategy of Pd-catalyzed intramolecular asymmetric aryl C–O coupling of 2-(2-halophenoxy)propane-1,3-diols, was developed. The SDP(O) ligand shows much better results than its SDP counterpart. The protocol provides an efficient and highly enantioselective method for the synthesis of 2-hydroxymethyl-1,4-benzodioxanes. Density functional theory studies provide a model that accounts for the origin of the enantioselectivity.
Co-reporter:Lin Zou, Xiao-Ye Wang, Xiao-Xiao Zhang, Ya-Zhong Dai, Yun-Dong Wu, Jie-Yu Wang and Jian Pei
Chemical Communications 2015 vol. 51(Issue 63) pp:12585-12588
Publication Date(Web):25 Jun 2015
DOI:10.1039/C5CC04860E
Electron-deficient pyrene-1,2,6,7-tetracarboxylic diimide (PyrDI) and its cyano derivative (PyrDI-CN) have been designed and synthesized as a new family of aromatic diimides. PyrDI has two unexpected five-membered imide rings and can form excimers facilely in the solid state. These new pyrene derivatives are promising n-type semiconductors for organic electronics.
Co-reporter:Chen-Yang Zhou, Fan Jiang, and Yun-Dong Wu
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 11) pp:5473-5480
Publication Date(Web):October 14, 2015
DOI:10.1021/acs.jctc.5b00581
To test whether our recently developed residue-specific force field RSFF2 can reproduce the mutational effect on the thermal stability of Trp-cage mini-protein and decipher its detailed folding mechanism, we carried out long-time replica-exchange molecular dynamics (REMD) simulations on five Trp-cage variants, including TC5b and TC10b. Initiated from their unfolded structures, the simulations not only well-reproduce their experimental structures but also their melting temperatures and folding enthalpies reasonably well. For each Trp-cage variant, the overall folding free energy landscape is apparently two-state, but some intermediate states can be observed when projected on more detailed coordinates. We also found different variants have the same major folding pathway, including the well formed PII-helix in the unfolded state, the formation of W6-P12/P18/P19 contacts and the α-helix before the transition state, the following formation of most native contacts, and the final native loop formation. The folding mechanism derived here is consistent with many previous simulations and experiments.
Co-reporter:Sangni Xun, Fan Jiang, and Yun-Dong Wu
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 4) pp:1949-1956
Publication Date(Web):March 3, 2015
DOI:10.1021/acs.jctc.5b00029
An important application of all-atom explicit-solvent molecular dynamics (MD) simulations is the refinement of protein structures from low-resolution experiments or template-based modeling. A critical requirement is that the native structure is stable with the force field. We have applied a recently developed residue-specific force field, RSFF1, to a set of 30 refinement targets from recent CASP experiments. Starting from their experimental structures, 1.0 μs unrestrained simulations at 298 K retain most of the native structures quite well except for a few flexible terminals and long internal loops. Starting from each homology model, a 150 ns MD simulation at 380 K generates the best RMSD improvement of 0.85 Å on average. The structural improvements roughly correlate with the RMSD of the initial homology models, indicating possible consistent structure refinement. Finally, targets TR614 and TR624 have been subjected to long-time replica-exchange MD simulations. Significant structural improvements are generated, with RMSD of 1.91 and 1.36 Å with respect to their crystal structures. Thus, it is possible to achieve realistic refinement of protein structure models to near-experimental accuracy, using accurate force field with sufficient conformational sampling.
Co-reporter:Chen-Yang Zhou, Fan Jiang, and Yun-Dong Wu
The Journal of Physical Chemistry B 2015 Volume 119(Issue 3) pp:1035-1047
Publication Date(Web):October 16, 2014
DOI:10.1021/jp5064676
Recently, we developed a residue-specific force field (RSFF1) based on conformational free-energy distributions of the 20 amino acid residues from a protein coil library. Most parameters in RSFF1 were adopted from the OPLS-AA/L force field, but some van der Waals and torsional parameters that effectively affect local conformational preferences were introduced specifically for individual residues to fit the coil library distributions. Here a similar strategy has been applied to modify the Amber ff99SB force field, and a new force field named RSFF2 is developed. It can successfully fold α-helical structures such as polyalanine peptides, Trp-cage miniprotein, and villin headpiece subdomain and β-sheet structures such as Trpzip-2, GB1 β-hairpins, and the WW domain, simultaneously. The properties of various popular force fields in balancing between α-helix and β-sheet are analyzed based on their descriptions of local conformational features of various residues, and the analysis reveals the importance of accurate local free-energy distributions. Unlike the RSFF1, which overestimates the stability of both α-helix and β-sheet, RSFF2 gives melting curves of α-helical peptides and Trp-cage in good agreement with experimental data. Fitting to the two-state model, RSFF2 gives folding enthalpies and entropies in reasonably good agreement with available experimental results.
Co-reporter:Fan Jiang
Journal of the American Chemical Society 2014 Volume 136(Issue 27) pp:9536-9539
Publication Date(Web):June 23, 2014
DOI:10.1021/ja502735c
Ab initio protein folding via physical-based all-atom simulation is still quite challenging. Using a recently developed residue-specific force field (RSFF1) in explicit solvent, we are able to fold a diverse set of 14 model proteins. The obtained structural features of unfolded state are in good agreement with previous observations. The replica-exchange molecular dynamics simulation is found to be efficient, resulting in multiple folding events for each protein. Transition path time is found to be significantly reduced under elevated temperature.
Co-reporter:Huan Sun, Chengming Wang, Yun-Fang Yang, Ping Chen, Yun-Dong Wu, Xinhao Zhang, and Yong Huang
The Journal of Organic Chemistry 2014 Volume 79(Issue 24) pp:11863-11872
Publication Date(Web):May 13, 2014
DOI:10.1021/jo500807d
Indole-containing polyaromatic scaffolds are widely found in natural products, pharmaceutical agents, and π-conjugated functional materials. Often, the synthesis of these highly valuable molecules requires a multistep sequence. Therefore, a simple, one-step protocol to access libraries of polyaromatic indole scaffolds is highly desirable. Herein we describe the direct synthesis of polysubstituted indolo[2,1-a]isoquinoline analogues via a double C–H annulation cascade using triazene as an internally cleavable directing group. Evidence from HRMS and theoretical calculations suggests that an unprecedented 1,2-alkyl migration might be responsible for the in situ cleavage of the directing group. Both kinetic isotope effects and DFT calculations suggested that the alkyne insertion step is rate-limiting for the second C,N annulation reaction.
Co-reporter:Yun-Fang Yang, Lung Wa Chung, Xinhao Zhang, K. N. Houk, and Yun-Dong Wu
The Journal of Organic Chemistry 2014 Volume 79(Issue 18) pp:8856-8864
Publication Date(Web):August 26, 2014
DOI:10.1021/jo501730n
Density functional theory calculations with the M06 functional have been performed on the reactivity, selectivity, and mechanism of hydrosilylations of alkynes, ketones, and nitriles catalyzed by cationic ruthenium complexes [CpRu(L)(MeCN)2]+, with L = PiPr3 or MeCN. The hydrosilylation of alkynes with L = PiPr3 involves an initial silyl migration mechanism to generate the anti-Markovnikov product, in contrast to the Markovnikov product obtained with L = MeCN. The bulky phosphine ligand directs the silyl group to migrate to Cβ of the alkyne. This explains the anti-Markovnikov selectivity of the catalyst with L = PiPr3. By contrast, the silane additions to either ketone or nitrile proceed through an ionic SN2-Si outer-sphere mechanism, in which the substrate attacks the Si center. The PiPr3 ligand facilitates the activation of the Si–H bond to furnish a η2-silane complex, whereas a η1-silane complex is formed for the MeCN ligand. This property of the phosphine ligand enables the catalytic hydrosilylation of ketones and nitriles in addition to that of alkynes.
Co-reporter:Fan Jiang, Chen-Yang Zhou, and Yun-Dong Wu
The Journal of Physical Chemistry B 2014 Volume 118(Issue 25) pp:6983-6998
Publication Date(Web):May 12, 2014
DOI:10.1021/jp5017449
Traditional protein force fields use one set of parameters for most of the 20 amino acids (AAs), allowing transferability of the parameters. However, a significant shortcoming is the difficulty to fit the Ramachandran plots of all AA residues simultaneously, affecting the accuracy of the force field. In this Feature Article, we report a new strategy for protein force field parametrization. Backbone and side-chain conformational distributions of all 20 AA residues obtained from protein coil library were used as the target data. The dihedral angle (torsion) potentials and some local nonbonded (1-4/1-5/1-6) interactions in OPLS-AA/L force field were modified such that the target data can be excellently reproduced by molecular dynamics simulations of dipeptides (blocked AAs) in explicit water, resulting in a new force field with AA-specific parameters, RSFF1. An efficient free energy decomposition approach was developed to separate the corrections on ϕ and ψ from the two-dimensional Ramachandran plots. RSFF1 is shown to reproduce the experimental NMR 3J-coupling constants of AA dipeptides better than other force fields. It has a good balance between α-helical and β-sheet secondary structures. It can successfully fold a set of α-helix proteins (Trp-cage and Homeodomain) and β-hairpins (Trpzip-2, GB1 hairpin), which cannot be consistently stabilized by other state-of-the-art force fields. Interestingly, the RSFF1 force field systematically overestimates the melting temperature (and the stability of native state) of these peptides/proteins. It has a potential application in the simulation of protein folding and protein structure refinement.
Co-reporter:Shengtao Ding ; Li-Juan Song ; Lung Wa Chung ; Xinhao Zhang ; Jianwei Sun
Journal of the American Chemical Society 2013 Volume 135(Issue 37) pp:13835-13842
Publication Date(Web):August 23, 2013
DOI:10.1021/ja405752w
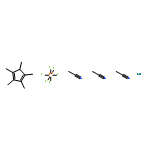
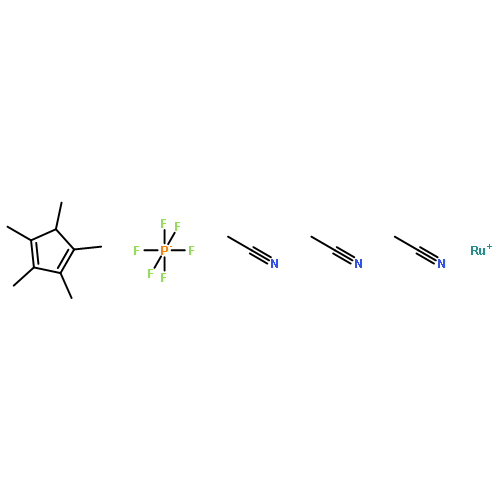
The first highly efficient ligand-controlled regio- and stereodivergent intermolecular hydrosilylations of internal alkynes have been disclosed. Cationic ruthenium complexes [Cp*Ru(MeCN)3]+ and [CpRu(MeCN)3]+ have been demonstrated to catalyze intermolecular hydrosilylations of silyl alkynes to form a range of vinyldisilanes with excellent but opposite regio- and stereoselectivity, with the former being α anti addition and the latter β syn addition. The use of a silyl masking group not only provides sufficient steric bulk for high selectivity but also leads to versatile product derivatizations toward a variety of useful building blocks. DFT calculations suggest that the reactions proceed by a mechanism that involves oxidative hydrometalation, isomerization, and reductive silyl migration. The energetics of the transition states and intermediates varies dramatically with the catalyst ligand (Cp* and Cp). Theoretical studies combined with experimental evidence confirm that steric effect plays a critical role in governing the regio- and stereoselectivity, and the interplay between the substituent in the alkyne (e.g., silyl group) and the ligand ultimately determines the observed remarkable regio- and stereodivergence.
Co-reporter:Gui-Juan Cheng ; Yun-Fang Yang ; Peng Liu ; Ping Chen ; Tian-Yu Sun ; Gang Li ; Xinhao Zhang ; K. N. Houk ; Jin-Quan Yu
Journal of the American Chemical Society 2013 Volume 136(Issue 3) pp:894-897
Publication Date(Web):December 30, 2013
DOI:10.1021/ja411683n
A combined experimental/computational study on the amino acid ligand-assisted Pd-catalyzed C–H bond activation reveals a mechanism in which the amino acid acts as both a dianionic bidentate ligand and a proton acceptor. This new model explains the effects of amino acids on reactivity and selectivity and unveils the dual roles of amino acids: stabilizing monomeric Pd complexes and serving as the internal base for proton abstraction.
Co-reporter:Gui-Juan Cheng;Li-Juan Song;Dr. Yun-Fang Yang;Dr. Xinhao Zhang;Dr. Olaf Wiest;Dr. Yun-Dong Wu
ChemPlusChem 2013 Volume 78( Issue 9) pp:943-951
Publication Date(Web):
DOI:10.1002/cplu.201300117
Abstract
A detailed computational study of a copper-catalyzed aerobic cross-dehydrogenative coupling reaction has been conducted. To select a reliable method to describe the thermochemistry of a single electron transfer (SET) process, benchmark calculations have been performed. M06/6-311+g(d,p) is appropriate to evaluate the thermochemistry of the SET process for the system involving iminium species. The computational results support an SET mechanism, but also uncover an alternative mechanism in which O2 is directly involved in a hydrogen-abstracting step. A comparative study with tert-butylhydroperoxide (TBHP) as the oxidant has also been performed. The computations reveal several competitive pathways, including a radical pathway, a CuIII pathway, and an SET mechanism for the Cu/TBHP system.
Co-reporter:Lin-tai Da and Yun-Dong Wu
Journal of Chemical Information and Modeling 2011 Volume 51(Issue 2) pp:359-369
Publication Date(Web):February 2, 2011
DOI:10.1021/ci1003448
The interaction between the HIV gp120 protein and coreceptor CCR5 or CXCR4 of the host cell is critical in mediating the HIV entry process. A model for the CCR5−gp120 complex has been developed. In the model, the N-terminus of CCR5 binds to three discontinuous domains of gp120, including the fourth conserved (C4) region, β19/β20 connecting loop, and V3 loop. The second extra-cellular loop (ECL2) of CCR5 also interacts with the crown part of the gp120 V3 loop. The bindings of the three CCR5 antagonists, maraviroc, aplaviroc, and vicriviroc, to the trans-membrane domain of CCR5 have been modeled. The bindings are found to affect the conformation of the ECL2 domain, which in turn drives the N-terminus of CCR5 to an altered state. Aplaviroc is more hydrophilic than maraviroc and vicriviroc, and its binding is more interfered by solvent, resulting in a quite different effect to the structure of CCR5 compared with those of the other two molecules. The above results are in accord with experimental observations and provide a structural basis for further design of CCR5 antagonists.
Co-reporter:Lin Zou, Xiao-Ye Wang, Xiao-Xiao Zhang, Ya-Zhong Dai, Yun-Dong Wu, Jie-Yu Wang and Jian Pei
Chemical Communications 2015 - vol. 51(Issue 63) pp:NaN12588-12588
Publication Date(Web):2015/06/25
DOI:10.1039/C5CC04860E
Electron-deficient pyrene-1,2,6,7-tetracarboxylic diimide (PyrDI) and its cyano derivative (PyrDI-CN) have been designed and synthesized as a new family of aromatic diimides. PyrDI has two unexpected five-membered imide rings and can form excimers facilely in the solid state. These new pyrene derivatives are promising n-type semiconductors for organic electronics.

![2-[(4s)-4-(2-methyl-2-propanyl)-4,5-dihydro-1,3-oxazol-2-yl]-5-(t Rifluoromethyl)pyridine](/data/chemimg/482100/1416819-91-4.png)
![2-[(4s)-4-(2-methyl-2-propanyl)-4,5-dihydro-1,3-oxazol-2-yl]-5-(t Rifluoromethyl)pyridine](/data/chemimg/482100/1416819-91-4_b.png)



![1-Methyl-6-phenyl-1H-imidazo[4,5-b]pyridin-2-amine](http://img.cochemist.com/ccimg/105700/105650-23-5.png)
![1-Methyl-6-phenyl-1H-imidazo[4,5-b]pyridin-2-amine](http://img.cochemist.com/ccimg/105700/105650-23-5_b.png)






![3-(4-Chlorophenyl)benzo[d]isoxazole](http://img.cochemist.com/ccimg/1227200/1227180-50-8.png)
![3-(4-Chlorophenyl)benzo[d]isoxazole](http://img.cochemist.com/ccimg/1227200/1227180-50-8_b.png)

![L-Proline,1-[(2-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/884700/884659-54-5.png)
![L-Proline,1-[(2-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/884700/884659-54-5_b.png)














![Cyclopropanecarboxylicacid, 1-[[(1,1-dimethylethoxy)carbonyl]amino]-2-phenyl-, (1S,2R)-](http://img.cochemist.com/ccimg/152000/151910-11-1.png)
![Cyclopropanecarboxylicacid, 1-[[(1,1-dimethylethoxy)carbonyl]amino]-2-phenyl-, (1S,2R)-](http://img.cochemist.com/ccimg/152000/151910-11-1_b.png)


![Benzene, [(butylthio)ethynyl]-](http://img.cochemist.com/ccimg/127100/127060-60-0.png)
![Benzene, [(butylthio)ethynyl]-](http://img.cochemist.com/ccimg/127100/127060-60-0_b.png)






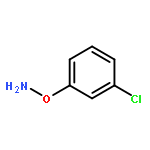
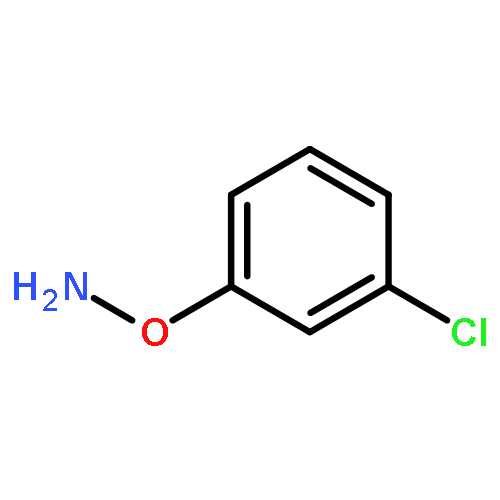




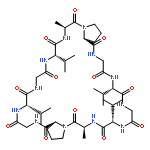
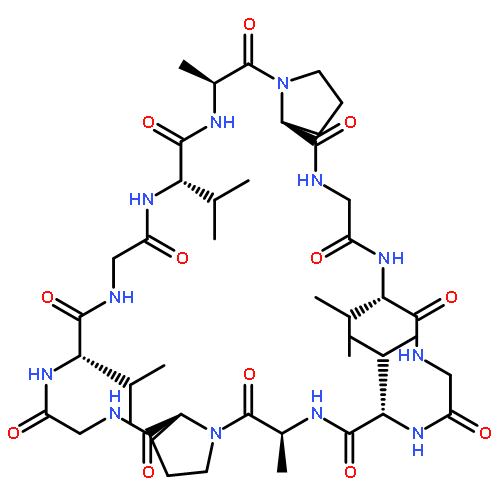
![L-Valine, N-[(2,2,2-trichloro-1,1-dimethylethoxy)carbonyl]-](http://img.cochemist.com/ccimg/66300/66270-38-0.png)
![L-Valine, N-[(2,2,2-trichloro-1,1-dimethylethoxy)carbonyl]-](http://img.cochemist.com/ccimg/66300/66270-38-0_b.png)















![Palladium,hexakis[m-(acetato-kO:kO')]tri-, cyclo](http://img.cochemist.com/ccimg/53200/53189-26-7.png)
![Palladium,hexakis[m-(acetato-kO:kO')]tri-, cyclo](http://img.cochemist.com/ccimg/53200/53189-26-7_b.png)









![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)









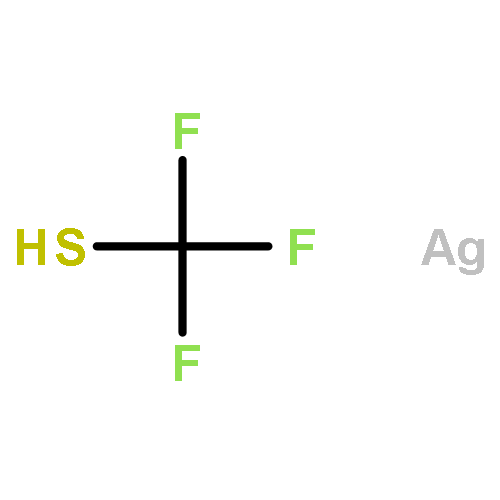
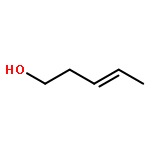
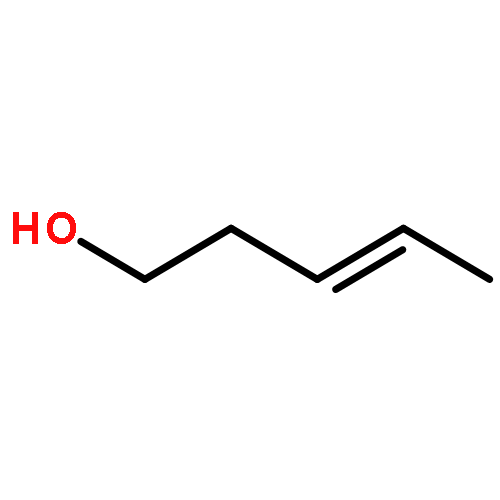


![Benzene, [(1-hexyn-1-ylthio)methyl]-](/data/chemimg/3765000/1355700-62-7.png)
![Benzene, [(1-hexyn-1-ylthio)methyl]-](/data/chemimg/3765000/1355700-62-7_b.png)