Co-reporter:Elizabeth A. Pillar and Marcelo I. Guzman
Environmental Science & Technology May 2, 2017 Volume 51(Issue 9) pp:4951-4951
Publication Date(Web):April 10, 2017
DOI:10.1021/acs.est.7b00232
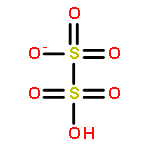
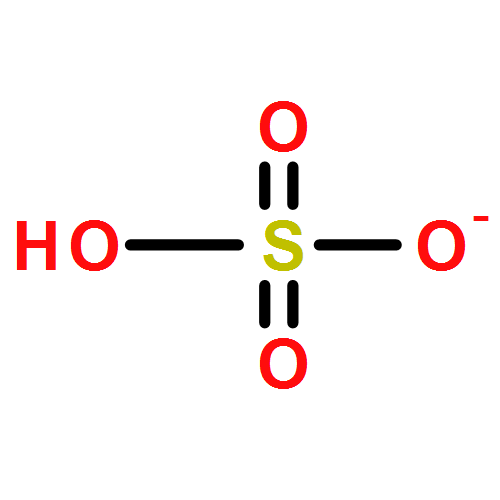
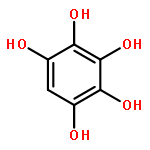
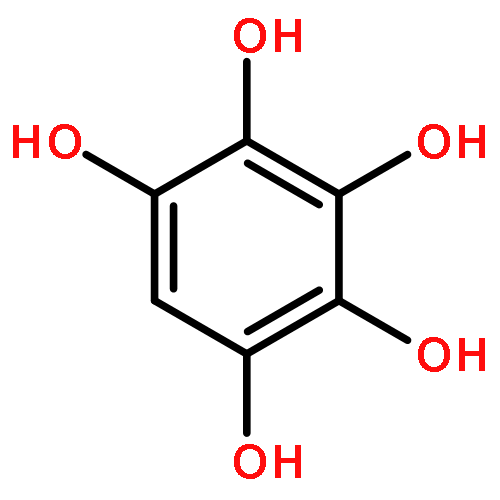
Anthropogenic activities contribute benzene, toluene, and anisole to the environment, which in the atmosphere are converted into the respective phenols, cresols, and methoxyphenols by fast gas-phase reaction with hydroxyl radicals (HO•). Further processing of the latter species by HO• decreases their vapor pressure as a second hydroxyl group is incorporated to accelerate their oxidative aging at interfaces and in aqueous particles. This work shows how catechol, pyrogallol, 3-methylcatechol, 4-methylcatechol, and 3-methoxycatechol (all proxies for oxygenated aromatics derived from benzene, toluene, and anisole) react at the air–water interface with increasing O3(g) during τc ≈ 1 μs contact time and contrasts their potential for electron transfer and in situ production of HO• using structure–activity relationships. A unifying mechanism is provided to explain the oxidation of the five proxies, which includes the generation of semiquinone radicals. Functionalization in the presence of HO• results in the formation of polyphenols and hydroxylated quinones. Instead, fragmentation produces polyfunctional low molecular weight carboxylic acids after oxidative cleavage of the aromatic bond with two vicinal hydroxy groups to yield substituted cis,cis-muconic acids. The generation of maleinaldehydic, maleic, pyruvic, glyoxylic, and oxalic acids confirms the potential of oxy aromatics to produce light-absorbing aqueous secondary organic aerosols in the troposphere.
Co-reporter:Alexis J. Eugene and Marcelo I. Guzman
The Journal of Physical Chemistry A April 20, 2017 Volume 121(Issue 15) pp:2924-2924
Publication Date(Web):March 31, 2017
DOI:10.1021/acs.jpca.6b11916
The variable composition of secondary organic aerosols (SOA) contributes to the large uncertainty for predicting radiative forcing. A better understanding of the reaction mechanisms leading to aerosol formation such as for the photochemical reaction of aqueous pyruvic acid (PA) at λ ≥ 305 nm can contribute to constrain these uncertainties. Herein, the photochemistry of aqueous PA (5–300 mM) continuously sparged with air is re-examined in the laboratory under comparable irradiance at 38° N at noon on a summer day. Several analytical methods are employed to monitor the time series of the reaction, including (1) the derivatization of carbonyl (C═O) functional groups with 2,4-dinitrophenylhydrazine (DNPH), (2) the separation of photoproducts by ultrahigh pressure liquid chromatography (UHPLC) and ion chromatography (IC) coupled to mass spectrometry (MS), (3) high resolution MS, (4) the assignment of 1H NMR and 13C gCOSY spectroscopic features, and (5) quantitative 1H NMR. The primary photoproducts are 2,3-dimethyltartaric acid and unstable 2-(1-carboxy-1-hydroxyethoxy)-2-methyl-3-oxobutanoic acid, a polyfunctional β-ketocarboxylic acid with eight carbons (C8) that quickly decarboxylates into 2-hydroxy-2-((3-oxobutan-2-yl)oxy)propanoic acid. Kinetic isotope effect studies performed for the first time for this system reveal the existence of tunneling during the initial loss of PA. Thus, the KIEs support a mechanism initiated by photoinduced proton coupled electron transfer (PCET). Measured reaction rates at variable initial [PA]0 were used to calculate the sum of the quantum yields for the products, which displays a hyperbolic dependence: ∑Φproduct = 1.99 [PA]0/(113.2 + [PA]0). The fast photochemical loss of aqueous PA with an estimated lifetime of 21.7 min is interpreted as a significant atmospheric sink for this species. The complexity of these aqueous phase pathways indicates that the solar photochemistry of an abundant α-ketocarboxylic acid can activate chemical processes for SOA formation.
Co-reporter:Matías E. Aguirre, Ruixin Zhou, Alexis J. Eugene, Marcelo I. Guzman, María A. Grela
Applied Catalysis B: Environmental 2017 Volume 217(Volume 217) pp:
Publication Date(Web):15 November 2017
DOI:10.1016/j.apcatb.2017.05.058
•A Cu2O/TiO2 type II heterostructure is characterized by XPS, DRUV, and UPS techniques.•The Cu2O/TiO2 type II heterostructure mediates CO2 photoreduction through a Z-scheme.•Detection of OH radicals only during UV–vis irradiation of Cu2O/TiO2 probes an operational Z-scheme.•Oxidation of Cu(I) during irradiation of Cu2O indicates the protective role of TiO2.The development of artificial photosynthesis aims to solve the increasing energy demand and associated environmental problems. A model photosynthetic system employing a composite of semiconductors with a Z-scheme can potentially mimic the combined power of photosystems 1 and 2 to transfer electrons. In this work, octahedral cuprous oxide covered with titanium dioxide nanoparticles (Cu2O/TiO2) are synthesized by a solvothermal strategy that provides high morphological and crystallographic control. The formation of a p-n heterojunction and characterization of the Type II band alignment of the composite are performed by diffuse reflectance UV-visible (DRUV) spectroscopy, ultraviolet photoelectron spectroscopy (UPS), and X-ray photoelectron spectroscopy (XPS). Upon UV-visible irradiation (λ ≥ 305 nm) of the composite in the presence of water vapor as the hole scavenger, the photoreduction of CO2(g) proceeds selectively to generate CO(g). The production rate of CO by the composite, RCO = 2.11 μmol gcat−1 h−1, is 4-times larger than for pure Cu2O under identical conditions. Contrasting XPS analyses of Cu2O and Cu2O/TiO2, during photocatalysts operation and the detection of photogenerated hydroxyl radicals (HO) in the heterostructure at variance with the results obtained for pure Cu2O are taken as evidences that TiO2 protects Cu2O from undergoing photocorrosion. These results provide direct evidence of an efficient Z-scheme as the main mechanism for harvesting energy during CO2 reduction in the synthesized materials.Download high-res image (187KB)Download full-size image
Co-reporter:Alexis J. Eugene, Sha-Sha Xia, and Marcelo I. Guzman
The Journal of Physical Chemistry A 2016 Volume 120(Issue 21) pp:3817-3826
Publication Date(Web):May 18, 2016
DOI:10.1021/acs.jpca.6b00225
Aerosols affect climate change, the energy balance of the atmosphere, and public health due to their variable chemical composition, size, and shape. While the formation of secondary organic aerosols (SOA) from gas phase precursors is relatively well understood, studying aqueous chemical reactions contributing to the total SOA budget is the current focus of major attention. Field measurements have revealed that mono-, di-, and oxo-carboxylic acids are abundant species present in SOA and atmospheric waters. This work explores the fate of one of these 2-oxocarboxylic acids, glyoxylic acid, which can photogenerate reactive species under solar irradiation. Additionally, the dark thermal aging of photoproducts is studied by UV–visible and fluorescence spectroscopies to reveal that the optical properties are altered by the glyoxal produced. The optical properties display periodicity in the time domain of the UV–visible spectrum of chromophores with absorption enhancement (thermochromism) or loss (photobleaching) during nighttime and daytime cycles, respectively. During irradiation, excited state glyoxylic acid can undergo α-cleavage or participate in hydrogen abstractions. The use of 13C nuclear magnetic resonance spectroscopy (NMR) analysis shows that glyoxal is an important intermediate produced during direct photolysis. Glyoxal quickly reaches a quasi-steady state as confirmed by UHPLC-MS analysis of its corresponding (E) and (Z) 2,4-dinitrophenylhydrazones. The homolytic cleavage of glyoxylic acid is proposed as a fundamental step for the production of glyoxal. Both carbon oxides, CO2(g) and CO(g) evolving to the gas-phase, are quantified by FTIR spectroscopy. Finally, formic acid, oxalic acid, and tartaric acid photoproducts are identified by ion chromatography (IC) with conductivity and electrospray (ESI) mass spectrometry (MS) detection and 1H NMR spectroscopy. A reaction mechanism is proposed based on all experimental observations.
Co-reporter:Ruixin Zhou
The Journal of Physical Chemistry C 2016 Volume 120(Issue 13) pp:7349-7357
Publication Date(Web):March 17, 2016
DOI:10.1021/acs.jpcc.5b12380
The reductive tricarboxylic acid (rTCA) cycle is an important central biosynthetic pathway that fixes CO2 into carboxylic acids. Among the five reductive steps in the rTCA cycle, the two-electron reduction of fumarate to succinate proceeds nonenzymatically on the surface of photoexcited sphalerite (ZnS) colloids suspended in water. This model reaction is chosen to systematically study the surface photoprocess occurring on ZnS in the presence of [Na2S] (1–10 mM) hole scavenger at 15 °C. Experiments at variable pH (5–10) indicate that monodissociated fumaric acid is the primary electron acceptor forming the monoprotic form of succinic acid. The following reaction scheme is proposed: (1) photoexcitation of ZnS generates conduction band electrons and valence band holes, (2) the hole scavenger donates electrons while producing sulfur-containing intermediates en route to sulfate formation, (3) a first electron transfer occurs at the conduction band converting chemisorbed monoprotic fumaric acid at surface zinc sites into an adsorb radical anion, and (4) the radical anion accepts a second electron and forms an adsorbed carbanion, which (5) abstracts two protons consecutively from either hydronium ion (acidic condition) or water (neutral and basic condition) to be desorbed as monodissociated succinic acid. The apparent quantum yield measurement of succinate production (Φs) under periodic irradiation at λ ≥ 305 nm shows that the time scale of electron transfer on the conduction band (t1) and valence band hole loss (t2) are in the order of hundred microseconds and a few milliseconds, respectively. These transitions (t1 and t2) become undistinguishable at 520 μs for a zeta potential ζ = −22.09 mV corresponding to [Na2S] = 0.57 mM. Overall, this work provides new insights to model heterogeneous processes such as the reduction of CO2 occurring on the surface of photocatalysts and advance present understanding of photocatalytic reactions.
Co-reporter:Elizabeth A. Pillar, Ruixin Zhou, and Marcelo I. Guzman
The Journal of Physical Chemistry A 2015 Volume 119(Issue 41) pp:10349-10359
Publication Date(Web):September 25, 2015
DOI:10.1021/acs.jpca.5b07914
Natural and anthropogenic emissions of aromatic hydrocarbons from biomass burning, agro-industrial settings, and fossil fuel combustion contribute precursors to secondary aerosol formation (SOA). How these compounds are processed under humid tropospheric conditions is the focus of current attention to understand their environmental fate. This work shows how catechol thin films, a model for oxygenated aromatic hydrocarbons present in biomass burning and combustion aerosols, undergo heterogeneous oxidation at the air–solid interface under variable relative humidity (RH = 0–90%). The maximum reactive uptake coefficient of O3(g) by catechol γO3 = (7.49 ± 0.35) × 10–6 occurs for 90% RH. Upon exposure of ca. 104-μm thick catechol films to O3(g) mixing ratios between 230 ppbv and 25 ppmv, three main reaction pathways are observed. (1) The cleavage of the 1,2 carbon–carbon bond at the air–solid interface resulting in the formation of cis,cis-muconic acid via primary ozonide and hydroperoxide intermediates. Further direct ozonolysis of cis,cis-muconic yields glyoxylic, oxalic, crotonic, and maleic acids. (2) A second pathway is evidenced by the presence of Baeyer–Villiger oxidation products including glutaconic 4-hydroxy-2-butenoic and 5-oxo-2-pentenoic acids during electrospray ionization mass spectrometry (MS) and ion chromatography MS analyses. (3) Finally, indirect oxidation by in situ produced hydroxyl radical (HO•) results in the generation of semiquinone radical intermediates toward the synthesis of polyhydoxylated aromatic rings such as tri-, tetra-, and penta-hydroxybenzene. Remarkably, heavier polyhydroxylated biphenyl and terphenyl products present in the extracted oxidized films result from coupling reactions of semiquinones of catechol and its polyhydroxylated rings. The direct ozonolysis of 1,2,3- and 1,2,4-trihydroxybenezene yields 2- and 3-hydroxy-cis,cis-muconic acid, respectively. The production of 2,4- or 3,4-dihdroxyhex-2-enedioic acid is proposed to result from the sequential processing of cis,cis-muconic acid, 2- and 3-hydroxy-cis,cis-muconic acid. Overall, these reactions contribute precursors to form aqueous SOA from aromatics in atmospheric aerosols and brown clouds.
Co-reporter:Elizabeth A. Pillar, Robert C. Camm, and Marcelo I. Guzman
Environmental Science & Technology 2014 Volume 48(Issue 24) pp:14352
Publication Date(Web):November 25, 2014
DOI:10.1021/es504094x
Anthropogenic emissions of aromatic hydrocarbons promptly react with hydroxyl radicals undergoing oxidation to form phenols and polyphenols (e.g., catechol) typically identified in the complex mixture of humic-like substances (HULIS). Because further processing of polyphenols in secondary organic aerosols (SOA) can continue mediated by a mechanism of ozonolysis at interfaces, a better understanding about how these reactions proceed at the air–water interface is needed. This work shows how catechol, a molecular probe of the oxygenated aromatic hydrocarbons present in SOA, can contribute interfacial reactive species that enhance the production of HULIS under atmospheric conditions. Reactive semiquinone radicals are quickly produced upon the encounter of 40 ppbv–6.0 ppmv O3(g) with microdroplets containing [catechol] = 1–150 μM. While the previous pathway results in the instantaneous formation of mono- and polyhydroxylated aromatic rings (PHA) and chromophoric mono- and polyhydroxylated quinones (PHQ), a different channel produces oxo- and dicarboxylic acids of low molecular weight (LMW). The cleavage of catechol occurs at the 1,2 carbon–carbon bond at the air–water interface through the formation of (1) an ozonide intermediate, (2) a hydroperoxide, and (3) cis,cis-muconic acid. However, variable [catechol] and [O3(g)] can affect the ratio of the primary products (cis,cis-muconic acid and trihydroxybenzenes) and higher order products observed (PHA, PHQ, and LMW oxo- and dicarboxylic acids). Secondary processing is confirmed by mass spectrometry, showing the production of crotonic, maleinaldehydic, maleic, glyoxylic, and oxalic acids. The proposed pathway can contribute precursors to aqueous SOA (AqSOA) formation, converting aromatic hydrocarbons into polyfunctional species widely found in tropospheric aerosols with light-absorbing brown carbon.
Co-reporter:Ruixin Zhou
The Journal of Physical Chemistry C 2014 Volume 118(Issue 22) pp:11649-11656
Publication Date(Web):May 12, 2014
DOI:10.1021/jp4126039
The photoreduction of CO2 to formate (HCOO–) in sphalerite (ZnS) aqueous suspensions is systematically studied in the presence of Na2S hole scavenger. A series of cut-on filters at λcut-on ≥ 280, 295, 305, 320, and 400 nm are used to measure the reaction rate of formate production. The dependence of the measured reaction rates on λcut-on indicates that a wavelength of λ = 345 nm is associated with the actual bandgap of the semiconductor nanocrystallites suspended in water. The results from apparent quantum yield measurements during periodic illumination experiments suggest that (1) valence-band holes on the surface of ZnS disappear within deciseconds due to the oxidation of the scavenger while simultaneously pumping electrons to the conduction band, (2) excited electrons in the conduction band of ZnS are transferred to CO2 to produce the intermediate CO2•–, and (3) CO2•– abstracts a proton from water and undergoes further photoreduction on the surface of ZnS in an overall time scale for steps 2 + 3 of a few milliseconds. The separation of both process merges at ∼29 ms because it decreases exponentially with a drop in [Na2S] accompanied by a less negative surface potential. The behavior of the reaction rate at variable pH resembles the fraction of dissolved CO2, discarding the direct participation of bicarbonate and carbonate in the reaction. Combined chromatographic, mass spectrometry, and spectroscopic studies provide new insights to understand the role of surface chemistry on the photoreduction of CO2 on ZnS nanocrystals.
Co-reporter:Elizabeth A. Pillar, Marcelo I. Guzman, and Jose M. Rodriguez
Environmental Science & Technology 2013 Volume 47(Issue 19) pp:10971-10979
Publication Date(Web):August 29, 2013
DOI:10.1021/es401700h
Halides are incorporated into aerosol sea spray, where they start the catalytic destruction of ozone (O3) over the oceans and affect the global troposphere. Two intriguing environmental problems undergoing continuous research are (1) to understand how reactive gas phase molecular halogens are directly produced from inorganic halides exposed to O3 and (2) to constrain the environmental factors that control this interfacial process. This paper presents a laboratory study of the reaction of O3 at variable iodide (I–) concentration (0.010–100 μM) for solutions aerosolized at 25 °C, which reveal remarkable differences in the reaction intermediates and products expected in sea spray for low tropospheric [O3]. The ultrafast oxidation of I– by O3 at the air–water interface of microdroplets is evidenced by the appearance of hypoiodous acid (HIO), iodite (IO2–), iodate (IO3–), triiodide (I3–), and molecular iodine (I2). Mass spectrometry measurements reveal an enhancement (up to 28%) in the dissolution of gaseous O3 at the gas–liquid interface when increasing the concentration of NaI or NaBr from 0.010 to 100 μM. The production of iodine species such as HIO and I2 from NaI aerosolized solutions exposed to 50 ppbv O3 can occur at the air–water interface of sea spray, followed by their transfer to the gas-phase, where they contribute to the loss of tropospheric ozone.
Co-reporter:Alexis J. Eugene;Sha-Sha Xia
PNAS 2013 110 (46 ) pp:E4274-E4275
Publication Date(Web):2013-11-12
DOI:10.1073/pnas.1313991110
Co-reporter:Marcelo I. Guzman, Richa R. Athalye, and Jose M. Rodriguez
The Journal of Physical Chemistry A 2012 Volume 116(Issue 22) pp:5428-5435
Publication Date(Web):May 16, 2012
DOI:10.1021/jp3011316
During the aerosolization process at the sea surface, halides are incorporated into aerosol droplets, where they may play an important role in tropospheric ozone chemistry. Although this process may significantly contribute to the formation of reactive gas phase molecular halogens, little is known about the environmental factors that control how halides selectively accumulate at the air–water interface. In this study, the production of sea spray aerosol is simulated using electrospray ionization (ESI) of 100 nM equimolar solutions of NaCl, NaBr, NaI, NaNO2, NaNO3, NaClO4, and NaIO4. The microdroplets generated are analyzed by mass spectrometry to study the comparative enrichment of anions (fX–) and their correlation with ion properties. Although no correlation exists between fX– and the limiting equivalent ionic conductivity, the correlation coefficient of the linear fit with the size of the anions RX–, dehydration free-energy ΔGdehyd, and polarizability α, follows the order: RX––2 > RX––1 > RX– > ΔGdehyd > α. The same pure physical process is observed in H2O and D2O. The factor fX– does not change with pH (6.8–8.6), counterion (Li+, Na+, K+, and Cs+) substitution effects, or solvent polarity changes in methanol– and ethanol–water mixtures (0 ≤ xH2O ≤ 1). Sodium polysorbate 20 surfactant is used to modify the structure of the interface. Despite the observed enrichment of I– on the air–water interface of equimolar solutions, our results of seawater mimic samples agree with a model in which the interfacial composition is increasingly enriched in I– < Br– < Cl– over the oceanic boundary layer due to concentration effects in sea spray aerosol formation.
.jpg)