Co-reporter:Ruirui Liu, Chenxi Zhang, Lingyan Kang, Xiaomin Sun and Yan Zhao
RSC Advances 2015 vol. 5(Issue 48) pp:37988-37994
Publication Date(Web):09 Apr 2015
DOI:10.1039/C5RA00612K
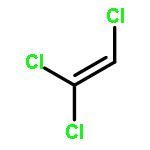
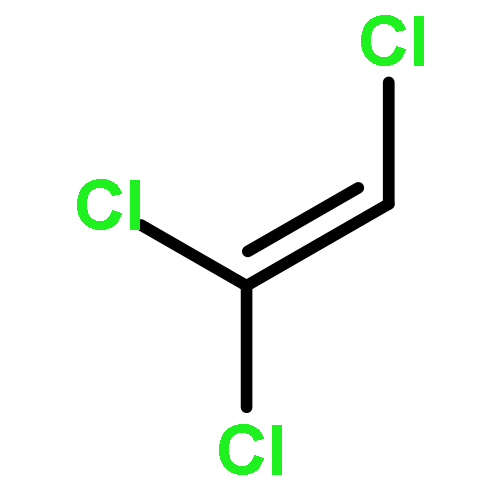
Short chain chlorinated paraffins (SCCPs) have recently drawn public attention because they have properties similar to persistent organic pollutants. In this study, 1,2,4,6,8,10,11-heptachloroundecane (HCU) is selected as a model molecule to investigate the chemical transformation of SCCPs using density functional theory (DFT) methods. After hydroxyl (OH) radicals initiate hydrogen (H) atom abstraction reactions, the produced intermediates could be further oxidized in the presence of O2/NO. The main products were found to be chloro-aldehydes and chloro-ketones after dechlorination reactions, or chloro-hydrins after reaction with H2O. These products have strong water solubility and polarity. Cl and OH radicals, which have strong reactivity, are also generated in the degradation process. Rate constants were calculated using transition state theory and the Arrhenius formulas were fitted. The total rate constant for the reaction of HCU with OH radicals is about 1.67 × 10−12 cm3 per molecule per s at room temperature. The atmospheric lifetime of HCU relative OH radicals is about 7.1 days.
Co-reporter:Xi Lu, Qian Liu, Xiaoyin Wang, Runjiao Cheng, Mingtao Zhang and Xiaomin Sun
RSC Advances 2015 vol. 5(Issue 4) pp:2827-2836
Publication Date(Web):11 Nov 2014
DOI:10.1039/C4RA12466A
A novel ruthenium complex developed by Casey exhibits some outstanding features such as effective anti-dimerization, highlighted catalytic activity and a mild reaction condition. Density functional theory (DFT) was used to explore the catalytic cycle of the hydrogenation of PhCHO catalyzed by this enhanced ruthenium complex. The catalytic cycle of aldehyde hydrogenation involves two stages, hydrogen transfer and regeneration of the active catalyst, which can be achieved by means of a concerted outer-sphere hydrogen transfer and an intramolecular hydrogen migration, respectively. Hydrogen transfer is the rate-determining step in the total catalytic hydrogenation cycle, having a low free energy barrier of 16.2 kcal mol−1. The hydrogenated product, alcohol, can remarkably improve the regeneration activity of the catalyst via an alcohol-mediated intramolecular hydrogen migration. The free energy barrier of regeneration of the active catalyst is only 13.8 kcal mol−1. This catalytic hydrogenation of aldehyde is demonstrated to be an autocatalytic process.
Co-reporter:Lingyan Kang, Chenxi Zhang and Xiaomin Sun
RSC Advances 2015 vol. 5(Issue 117) pp:96518-96524
Publication Date(Web):05 Nov 2015
DOI:10.1039/C5RA11453E
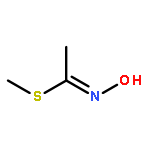
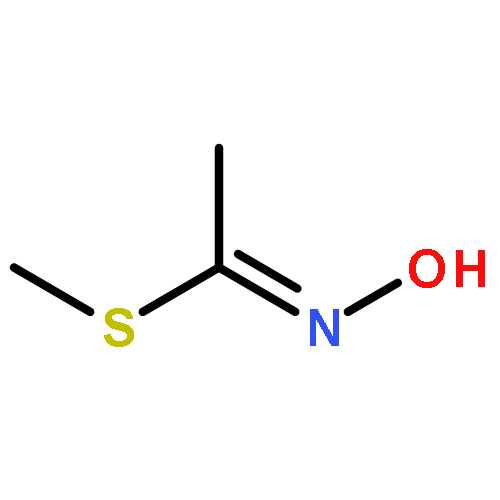
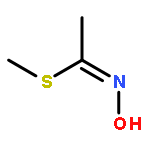
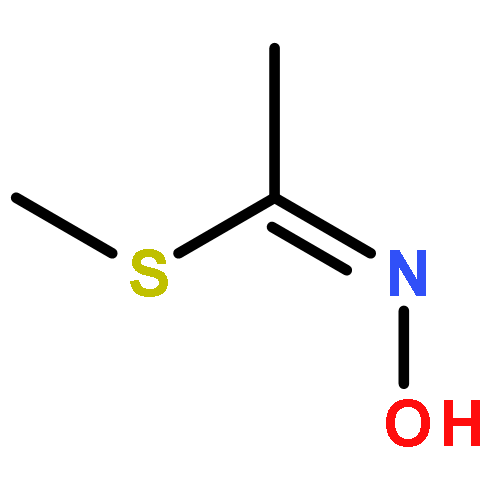
Acetofenate (AF) is a widely used pesticide. The mechanism of HOx, NO3, O3, and Cl initiated oxidation reactions of AF was investigated with density functional theory. For each of the OH, NO3, and Cl, both addition and hydrogen abstraction were investigated, for each of the HO2 radicals and O3, addition reactions were investigated. The cycloaddition reactions of O3 were considered, including the exploration of isomerization. Based on the potential energy surface, the rate constants were calculated with the transition state theory method over a temperature range of 200–400 K and fitted with the Arrhenius formulas. The rate constants of the AF reaction with OH, HO2, NO3, O3, and Cl, are 4.04 × 10−13, 7.02 × 10−33, 6.93 × 10−20, 1.45 × 10−25, and 5.07 × 10−12 cm3 per molecule per s at 298.15 K, respectively. The OH-initiated reactions are dominant according to the branching ratio of reaction constants. The atmospheric lifetime of the reaction species was estimated according to rate constants.
Co-reporter:Xi Lu, Runjiao Cheng, Nicholas Turner, Qian Liu, Mingtao Zhang, and Xiaomin Sun
The Journal of Organic Chemistry 2014 Volume 79(Issue 19) pp:9355-9364
Publication Date(Web):September 15, 2014
DOI:10.1021/jo501946k
Knölker’s iron complex is a “green” catalyst that exhibits low toxicity and is abundant in nature. Density functional theory (DFT) was used to explore the highly chemoselective nature of the catalytic hydrogenation of CH2═CHCH2CHO. An outer-sphere concerted hydrogen transfer was found to be the most reasonable kinetic route for the hydrogenation of the olefin. However, the C═C hydrogenation reaction has a high free energy barrier of 28.1 kcal/mol, requiring a high temperature to overcome. By comparison, the CH═O bond concerted hydrogen-transfer reaction catalyzed using Knölker’s iron catalyst has an energy barrier of only 14.0 kcal/mol. Therefore, only the CH═O of CH2═CHCH2CHO can be hydrogenated in the presence of Knölker’s catalyst at room temperature, due to kinetic domination. All computational results were in good agreement with experimental results.
Co-reporter:Chenxi Zhang, Tingli Sun, and Xiaomin Sun
Environmental Science & Technology 2011 Volume 45(Issue 11) pp:4756-4762
Publication Date(Web):May 3, 2011
DOI:10.1021/es104271a
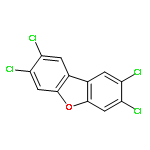
The atmospheric oxidation of 2,3,7,8-tetrachlorodibenzo-p-dioxin (2,3,7,8-TeCDD) is investigated theoretically by high-accuracy molecular orbital calculation. The study shows that the OH radical can easily be added to the C atom adjacent to the O atom in dioxin ring to form OH radical adduct. The 2,3,7,8-TeCDD–OH adduct can immediately react with O2 to form the 2,3,7,8-TeCDD–OH-O2 adduct which can react with NO or H2O to complete the decomposition process. The degradation mechanism varies with the addition position of O2 and the O-abstraction by NO. The OH radical can be reproduced through the H-abstraction of H2O and initiate a new round of degradation. The direct dynamic calculation is performed, and the rate constants is calculated over a temperature range of 200–1200 K, using the canonical variational transition state theory with small-curvature tunneling effect. The four-parameter formula of rate constants with the temperature is fitted and the lifetimes of the reaction species in the troposphere are estimated according to the rate constants, which is helpful for the atmospheric model study on the formation and degradation of dioxin.
Co-reporter:Chenxi Zhang, Xiaomin Sun, Yisheng Xu, Chuansong Qi, Jianghua Zhang
Journal of Environmental Chemical Engineering (June 2014) Volume 2(Issue 2) pp:1098-1103
Publication Date(Web):1 June 2014
DOI:10.1016/j.jece.2014.04.012
•Atmospheric reactions of TCDD/F initiated by OH and HO2, O3, NO3, and Cl were investigated.•TCDD/F will be mainly scavenged by OH during daytime, while NO3 is the primary oxidant at night.•Reactions with Cl atoms are the dominant removal pathway of TCDD/F in coastal area after sunrise.The reactions of gas-phase PCDD/Fs with oxidizing agents are believed to be the major atmospheric sink. In this study, the density functional theory (DFT) method is employed to investigate the reaction mechanisms of 2,3,7,8-tetrachlorinated dibenzo-p-dioxins (2,3,7,8-TCDD) and 2,3,7,8-tetra chlorinated dibenzofuran (2,3,7,8-TCDF) initiated by a series of atmospheric oxidants (e.g., OH, HO2, O3, NO3 radicals, and Cl atom). The rate constants obtained from the reactions of the 2,3,7,8-TCDD/F with the corresponding oxidants are 7.75 × 10−12/9.78 × 10−13, 2.53 × 10−25/3.61 × 10−25, 3.71 × 10−21/6.86 × 10−22, 3.96 × 10−14/6.46 × 10−15, and 1.23 × 10−9/1.37 × 10−10 cm3 molecule−1 s−1, respectively. The results show that 2,3,7,8-TCDD/F will be mainly scavenged by OH radical, while NO3 radical is the primary oxidant at night and Cl atom dominates after sunrise in coastal and marine areas.
Co-reporter:Lin He, Xiaomin Sun, Fanping Zhu, Shaojie Ren, Shuguang Wang
Science of The Total Environment (15 August 2017) Volume 592() pp:33-40
Publication Date(Web):15 August 2017
DOI:10.1016/j.scitotenv.2017.03.041
•OH-initiated transformation of aspirin in aqueous environments was investigated.•H-abstraction pathways at methyl position OH-addition pathways were dominated rather than hydrolysis reactions in AOPs.•Hydrolysis mechanisms of organic ester compound were investigated by DFT methods for the first time.•UV/H2O2 experiments were performed to confirm calculation findings.Advanced oxidation processes (AOPs) are widely used in wastewater treatment of pharmaceutical and personal care products (PPCPs). In this work, the OH-initiated transformation as well as the hydrolysis of a typical PPCPs, aspirin, was investigated using density functional theory (DFT) calculations and laboratory experiments. For DFT calculations, the frontier electron densities and bond dissociation energies were analyzed. Profiles of the potential energy surface were constructed, and all the possible pathways were discussed. Additionally, rate constants for each pathway were calculated with transition state theory (TST) method. UV/H2O2 experiments of aspirin were performed and degradation intermediates were identified by UPLC-MS-MS analysis. Different findings from previous experimental works were reported that the H-abstraction pathways at methyl position were dominated and OH-addition pathways on benzene ring were also favored. Meantime, hydroxyl ASA was confirmed as the main stable intermediate. Moreover, it was the first time to use DFT method to investigate the hydrolysis mechanisms of organic ester compound.Download high-res image (95KB)Download full-size image





















![2,3,7,8-Tetrachlorodibenzo[b,e][1,4]dioxine](http://img.cochemist.com/ccimg/1800/1746-01-6.png)
![2,3,7,8-Tetrachlorodibenzo[b,e][1,4]dioxine](http://img.cochemist.com/ccimg/1800/1746-01-6_b.png)

