Co-reporter:Barbara Guerra, Nils Bischoff, Volodymyr G. Bdzhola, Sergiy M. Yarmoluk, Olaf-Georg Issinger, Andriy G. Golub, and Karsten Niefind
ACS Chemical Biology 2015 Volume 10(Issue 7) pp:1654
Publication Date(Web):May 11, 2015
DOI:10.1021/acschembio.5b00235
CK2 is a Ser/Thr kinase recruited by tumor cells to avoid cell death. 4′-Carboxy-6,8-dibromo-flavonol (FLC26) is a nanomolar CK2 inhibitor reducing the physiological phosphorylation of CK2 biomarkers and inducing cell death. Its binding mode to the ATP site was predicted to depend primarily on noncovalent interactions not comprising halogen bonds. We confirm this by two independent cocrystal structures which additionally show that FLC26 is selective for an open, protein kinase-untypical conformation of the hinge/helix αD region. The structures suggest how the bromo substituents, found previously in lead optimization studies, contribute to the inhibitory efficacy. In this context, one of the complex structures, obtained by crystallization with the kosmotropic salt NaCl, revealed an unconventional π-halogen bond between the 8-bromo substituent of FLC26 and an aromatic side chain which is absent under low-salt conditions. The kosmotropic salt sensitivity of π-halogen bonds is a novel feature which requires attention in structural comparisons and halogen-bond-based explanations.
Co-reporter:Jennifer Hochscherf, Dirk Lindenblatt, Michaela Steinkrüger, Eungyoung Yoo, Özlem Ulucan, Stefan Herzig, Olaf-Georg Issinger, Volkhard Helms, Claudia Götz, Ines Neundorf, Karsten Niefind, Markus Pietsch
Analytical Biochemistry 2015 Volume 468() pp:4-14
Publication Date(Web):1 January 2015
DOI:10.1016/j.ab.2014.09.003
Abstract
Increased activity of protein kinase CK2 is associated with various types of cancer, neurodegenerative diseases, and chronic inflammation. In the search for CK2 inhibitors, attention has expanded toward compounds disturbing the interaction between CK2α and CK2β in addition to established active site-directed approaches. The current article describes the development of a fluorescence anisotropy-based assay that mimics the principle of CK2 subunit interaction by using CK2α1–335 and the fluorescent probe CF-Ahx-Pc as a CK2β analog. In addition, we identified new inhibitors able to displace the fluorescent probe from the subunit interface on CK2α1–335. Both CF-Ahx-Pc and the inhibitors I-Pc and Cl-Pc were derived from the cyclic peptide Pc, a mimetic of the C-terminal CK2α-binding motif of CK2β. The design of the two inhibitors was based on docking studies using the known crystal structure of the Pc/CK2α1–335 complex. The dissociation constants obtained in the fluorescence anisotropy assay for binding of all compounds to human CK2α1–335 were validated by isothermal titration calorimetry. I-Pc was identified as the tightest binding ligand with a KD value of 240 nM and was shown to inhibit the CK2 holoenzyme-dependent phosphorylation of PDX-1, a substrate requiring the presence of CK2β, with an IC50 value of 92 μM.
Co-reporter:Jennifer Raaf, Barbara Guerra, Ines Neundorf, Bertan Bopp, Olaf-Georg Issinger, Joachim Jose, Markus Pietsch, and Karsten Niefind
ACS Chemical Biology 2013 Volume 8(Issue 5) pp:901
Publication Date(Web):March 8, 2013
DOI:10.1021/cb3007133
The constitutively active Ser/Thr kinase CK2 (casein kinase 2) is used by tumor cells to acquire apoptosis resistance. CK2 exists as a heterotetrameric holoenzyme with two catalytic chains (CK2α) attached to a dimer of noncatalytic subunits (CK2β). A druggable cavity at the CK2β interface of CK2α allows the design of small molecules disturbing the CK2α/CK2β interaction and thus affecting activity, stability, and substrate specificity. We describe here the first structure of CK2α with an effective CK2β-competitive compound, namely, a 13-meric cyclic peptide derived from the C-terminal CK2β segment. Some well-ordered water molecules not visible in CK2 holoenzyme structures were detected at the interface. Driven mainly by enthalpy, the peptide binds with submicromolar affinity to CK2α, stimulates its catalytic activity, and reduces effectively the CK2α/CK2β affinity. The results provide a thermodynamic and structural rationalization of the peptide’s CK2β-competitive functionality and pave thus the way to a peptidomimetic drug addressing the CK2α/CK2β interaction.
Co-reporter:
Biochemistry 2011 Volume 50(Issue 4) pp:512-522
Publication Date(Web):December 13, 2010
DOI:10.1021/bi1013563
The protein Ser/Thr kinase CK2 (former name: casein kinase II) exists predominantly as a heterotetrameric holoenzyme composed of two catalytic subunits (CK2α) bound to a dimer of noncatalytic subunits (CK2β). We undertook a study to further understand how these subunits interact to form the tetramer. To this end, we used recombinant, C-terminal truncated forms of human CK2 subunits that are able to form the holoenzyme. We analyzed the interaction thermodynamics between the binding of CK2α and CK2β as well as the impact of changes in temperature, pH, and the ionization enthalpy of the buffer using isothermal titration calorimetry (ITC). With structure-guided alanine scanning mutagenesis we truncated individual side chains in the hydrophobic amino acid cluster located within the CK2α interface to identify experimentally the amino acids that dominate affinity. The ITC results indicate that Leu41 or Phe54 single mutations were most disruptive to binding of CK2β. Additionally, these CK2α mutants retained their kinase activity. Furthermore, the substitution of Leu41 in combination with Phe54 showed that the individual mutations were not additive, suggesting that the cooperative action of both residues played a role. Interestingly, the replacement of Ile69, which has a central position in the interaction surface of CK2α, only had modest effects. The differences between Leu41, Phe54, and Ile69 in interaction relevance correlate with solvent accessibility changes during the transition from unbound to CK2β-bound CK2α. Identifying residues on CK2α that play a key role in CK2α/CK2β interactions is important for the future generation of small molecule drug design.
Co-reporter:Karsten Niefind, Olaf-Georg Issinger
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2010 Volume 1804(Issue 3) pp:484-492
Publication Date(Web):March 2010
DOI:10.1016/j.bbapap.2009.09.022
At the first glance CK2α, the catalytic subunit of protein kinase CK2, is a rigid molecule: in contrast to many eukaryotic protein kinases in CK2α the canonical regulatory key elements like the activation segment occur exclusively in their typical active conformations. This observation fits well to the constitutive activity of the enzyme, meaning, its independence from phosphorylation or other characteristic control factors. Most CK2α structures are based on the enzyme from Zea mays, supplemented by an increasing number of human CK2α structures. In the latter a surprising plasticity of important ATP-binding elements – the interdomain hinge region and the glycine-rich loop – was discovered. In fully active CK2α the hinge region is open and does not anchor the ATP ribose, but alternatively it can adopt a closed conformation, form hydrogen bonds to the ribose moiety and thus retract the γ-phospho group from its functional position. In addition to this partially inactive state human CK2α was recently found in a fully inactive conformation. It is incompatible with ATP-binding due to a combination of a closed hinge and a collapse of the glycine-rich loop into the ATP cavity. These conformational transitions are apparently correlated with the occupation state of a remote docking site located at the interface to the non-catalytic subunit CK2β: if CK2β blocks this site, the fully active conformation of CK2α is stabilized, while the binding of certain small molecule seems to favour the partially and fully inactive states. This observation may be exploited to design effective and selective CK2 inhibitors.
Co-reporter:K. Niefind;J. Raaf;O.-G. Issinger
Cellular and Molecular Life Sciences 2009 Volume 66( Issue 11-12) pp:1800-1816
Publication Date(Web):2009 June
DOI:10.1007/s00018-009-9149-8
Within the last decade, 40 crystal structures corresponding to protein kinase CK2 (former name ‘casein kinase 2’), to its catalytic subunit CK2α and to its regulatory subunit CK2β were published. Together they provide a valuable, yet by far not complete basis to rationalize the biochemical features of the enzyme, such as its constitutive activity, acidophilic substrate specificity, dual-cosubstrate specificity and its heterotetrameric quarternary structure. Comprehensive sets of structural superimpositions reveal that both CK2α and CK2β are relatively rigid molecules. In CK2β the critical region of CK2α recruitment is pre-formed in the unbound state. In CK2α the activation segment – a key element of protein kinase regulation – adapts invariably the typical conformation of the active enzymes. Recent structures of human CK2α revealed a surprising plasticity in the ATP-binding region, suggesting an alternative mode of activity control.
Co-reporter:K. Niefind;J. Raaf;O.-G. Issinger
Cellular and Molecular Life Sciences 2009 Volume 66( Issue 21) pp:3535-3536
Publication Date(Web):2009 November
DOI:10.1007/s00018-009-0128-x
Co-reporter:Jennifer Raaf, Elena Brunstein, Olaf-Georg Issinger, Karsten Niefind
Chemistry & Biology 2008 Volume 15(Issue 2) pp:111-117
Publication Date(Web):22 February 2008
DOI:10.1016/j.chembiol.2007.12.012
The Ser/Thr kinase CK2 (previously called casein kinase 2) is composed of two catalytic chains (CK2α) attached to a dimer of noncatalytic subunits (CK2β). CK2 is involved in suppression of apoptosis, cell survival, and tumorigenesis. To investigate these activities and possibly affect them, selective CK2 inhibitors are required. An often-used CK2 inhibitor is 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB). In a complex structure with human CK2α, DRB binds to the canonical ATP cleft, but additionally it occupies an allosteric site that can be alternatively filled by glycerol. Inhibition kinetic studies corroborate the dual binding mode of the inhibitor. Structural comparisons reveal a surprising conformational plasticity of human CK2α around both DRB binding sites. After local rearrangement, the allosteric site serves as a CK2β interface. This opens the potential to construct molecules interfering with the CK2α/CK2β interaction.
Co-reporter:Jennifer Raaf;Olaf-Georg Issinger
Molecular and Cellular Biochemistry 2008 Volume 316( Issue 1-2) pp:15-23
Publication Date(Web):2008 September
DOI:10.1007/s11010-008-9826-1
The diffraction pattern of a protein crystal is normally a product of the interference of electromagnetic waves scattered by electrons of the crystalline sample. The diffraction pattern undergoes systematic changes in case additionally X-ray absorption occurs, meaning if the wavelength of the primary X-ray beam is relatively close to the absorption edge of selected elements of the sample. The resulting effects are summarized as “anomalous dispersion” and can be always observed with “soft” X-rays (wavelength around 2 Å) since they match the absorption edges of sulfur and chlorine. A particularly useful application of this phenomenon is the experimental detection of the sub-structures of the anomalous scatterers in protein crystals. We demonstrate this here with a crystal of a C-terminally truncated variant of human CK2α to which two molecules of the inhibitor 5,6-dichloro-1-β-d-ribo-furanosyl-benzimidazole (DRB) are bound. The structure of this co-crystal has been solved recently. For this study we measured an additional diffraction data set at a wavelength of 2 Å which showed strong anomalous dispersion effects. On the basis of these effects we detected all sulfur atoms of the protein, the two liganded DRB molecules and a total of 16 additional chloride ions some of them emerging at positions filled with water molecules in previous structure determinations. A number of chloride ions are bound to structural and functional important locations fitting to the constitutive activity and the acidophilic substrate specificity of the enzyme.
Co-reporter:Nina Richter, Klaus Breicha, Werner Hummel, Karsten Niefind
Journal of Molecular Biology (3 December 2010) Volume 404(Issue 3) pp:353-362
Publication Date(Web):3 December 2010
DOI:10.1016/j.jmb.2010.09.049
The NADP-dependent glycerol dehydrogenase (EC 1.1.1.72) from Gluconobacter oxydans is a member of family 11 of the aldo–keto reductase (AKR) enzyme superfamily; according to the systematic nomenclature within the AKR superfamily, the term AKR11B4 has been assigned to the enzyme. AKR11B4 is a biotechnologically attractive enzyme because of its broad substrate spectrum, combined with its distinctive regioselectivity and stereoselectivity. These features can be partially rationalized based on a 2-Å crystal structure of apo-AKR11B4, which we describe and interpret here against the functional complex structures of other members of family 11 of the AKR superfamily. The structure of AKR11B4 shows the AKR-typical (β/α)8 TIM-barrel fold, with three loops and the C-terminal tail determining the particular enzymatic properties. In comparison to AKR11B1 (its closest AKR relative), AKR11B4 has a relatively broad binding cleft for the cosubstrate NADP/NADPH. In the crystalline environment, it is completely blocked by the C-terminal segment of a neighboring protomer. The structure reveals a conspicuous tryptophan residue (Trp23) that has to adopt an unconventional and strained side-chain conformation to permit cosubstrate binding. We predict and confirm by site-directed mutagenesis that Trp23 is an accelerator of (co)substrate turnover. Furthermore, we show that, simultaneously, this tryptophan residue is a critical determinant for substrate binding by the enzyme, while enantioselectivity is probably governed by a methionine residue within the C-terminal tail. We present structural reasons for these notions based on ternary complex models of AKR11B4, NADP, and either octanal, d-glyceraldehyde, or l-glyceraldehyde.Download high-res image (194KB)Download full-size image
Co-reporter:Guido Hansen, Heike Gielen-Haertwig, Peter Reinemer, Dietmar Schomburg, ... Karsten Niefind
Journal of Molecular Biology (24 June 2011) Volume 409(Issue 5) pp:681-691
Publication Date(Web):24 June 2011
DOI:10.1016/j.jmb.2011.04.047
Human neutrophil elastase (HNE), a trypsin-type serine protease, is of pivotal importance in the onset and progression of chronic obstructive pulmonary disease (COPD). COPD encompasses a group of slowly progressive respiratory disorders and is a major medical problem and the fifth leading cause of death worldwide. HNE is a major target for the development of compounds that inhibit the progression of long-term lung function decline in COPD patients.Here, we present the three-dimensional structure of a potent dihydropyrimidone inhibitor (DHPI) non-covalently bound to HNE at a resolution of 2.0 Å. The inhibitor binds to the active site in a unique orientation addressing S1 and S2 subsites of the protease. To facilitate further analysis of this binding mode, we determined the structure of the uncomplexed enzyme at a resolution of 1.86 Å. Detailed comparisons of the HNE:DHPI complex with the uncomplexed HNE structure and published structures of other elastase:inhibitor complexes revealed that binding of DHPI leads to large conformational changes in residues located in the S2 subsite. The rearrangement of residues Asp95–Leu99B creates a deep, well-defined cavity, which is filled by the P2 moiety of the inhibitor molecule to almost perfect shape complementarity. The shape of the S2 subsite in complex with DHPI clearly differs from all other observed HNE structures. The observed structural flexibility of the S2 subsite is a key feature for the understanding of the binding mode of DHPIs in general and the development of new HNE selective inhibitors.Graphical AbstractDownload high-res image (227KB)Download full-size image
Co-reporter:Nils Bischoff, Birgitte Olsen, Jennifer Raaf, Maria Bretner, ... Karsten Niefind
Journal of Molecular Biology (18 March 2011) Volume 407(Issue 1) pp:1-12
Publication Date(Web):18 March 2011
DOI:10.1016/j.jmb.2011.01.020
Protein kinase CK2 (formerly “casein kinase 2”) is composed of a central dimer of noncatalytic subunits (CK2β) binding two catalytic subunits. In humans, there are two isoforms of the catalytic subunit (and an additional splicing variant), one of which (CK2α) is well characterized. To supplement the limited biochemical knowledge about the second paralog (CK2α′), we developed a well-soluble catalytically active full-length mutant of human CK2α′, characterized it by Michaelis–Menten kinetics and isothermal titration calorimetry, and determined its crystal structure to a resolution of 2 Å. The affinity of CK2α′ for CK2β is about 12 times lower than that of CK2α and is less driven by enthalpy. This result fits the observation that the β4/β5 loop, a key element of the CK2α/CK2β interface, adopts an open conformation in CK2α′, while in CK2α, it opens only after assembly with CK2β. The open β4/β5 loop in CK2α′ is stabilized by two elements that are absent in CK2α: (1) the extension of the N-terminal β-sheet by an additional β-strand, and (2) the filling of a conserved hydrophobic cavity between the β4/β5 loop and helix αC by a tryptophan residue. Moreover, the interdomain hinge region of CK2α′ adopts a fully functional conformation, while unbound CK2α is often found with a nonproductive hinge conformation that is overcome only by CK2β binding. Taken together, CK2α′ exhibits a significantly lower affinity for CK2β than CK2α; moreover, in functionally critical regions, it is less dependent on CK2β to obtain a fully functional conformation.Graphical AbstractDownload high-res image (216KB)Download full-size image
Co-reporter:Jennifer Raaf, Karsten Klopffleisch, Olaf-Georg Issinger, Karsten Niefind
Journal of Molecular Biology (14 March 2008) Volume 377(Issue 1) pp:1-8
Publication Date(Web):14 March 2008
DOI:10.1016/j.jmb.2008.01.008
The Ser/Thr kinase CK2 (former name: casein kinase 2) is a heterotetrameric enzyme composed of two catalytic chains (CK2α) attached to a dimer of noncatalytic subunits. Together with the cyclin-dependent kinases and the mitogen-activated protein kinases, CK2α belongs to the CMGC family of the eukaryotic protein kinases. CK2 is an important survival and stability factor in eukaryotic cells: its catalytic activity is elevated in a wide variety of tumors while its down-regulation can lead to apoptosis. Thus, CK2 is a valuable target for drug development and for chemical biology approaches of cell biological research, and small organic inhibitors addressing CK2 are of considerable interest. We describe here the complex structure between a C-terminal deletion mutant of human CK2α and the ATP-competitive inhibitor emodin (1,3,8-trihydroxy-6-methylanthraquinone, International Union of Pure and Applied Chemistry name: 1,3,8-trihydroxy-6-methylanthracene-9,10-dione) and compare it with a previously published complex structure of emodin and maize CK2α. With a resolution of 1.5 Å, the human CK2α/emodin structure has a much better resolution than its maize counterpart (2.6 Å). Even more important, in spite of a sequence identity of more than 77% between human and maize CK2α, the two structures deviate significantly in the orientation, in which emodin is trapped by the enzyme, and in the local conformations around the ligand binding site: maize CK2α shows its largest adaptations in the ATP-binding loop, whereas human CK2α shows its largest adaptations in the hinge region connecting the two main domains of the protein kinase core. These observations emphasize the importance of local plasticity for ligand binding and demonstrate that two orthologues of an enzyme can behave quite different in this respect.
Co-reporter:Jennifer Raaf, Olaf-Georg Issinger, Karsten Niefind
Journal of Molecular Biology (13 March 2009) Volume 386(Issue 5) pp:1212-1221
Publication Date(Web):13 March 2009
DOI:10.1016/j.jmb.2009.01.033
The Ser/Thr kinase casein kinase 2 (CK2) is a heterotetrameric enzyme composed of two catalytic chains (CK2α, catalytic subunit of CK2) attached to a dimer of two noncatalytic subunits (CK2β, noncatalytic subunit of CK2). CK2α belongs to the superfamily of eukaryotic protein kinases (EPKs). To function as regulatory key components, EPKs normally exist in inactive ground states and are activated only upon specific signals. Typically, this activation is accompanied by large conformational changes in helix αC and in the activation segment, leading to a characteristic arrangement of catalytic key elements. For CK2α, however, no strict physiological control of activity is known. Accordingly, CK2α was found so far exclusively in the characteristic conformation of active EPKs, which is, in this case, additionally stabilized by a unique intramolecular contact between the N-terminal segment on one side, and helix αC and the activation segment on the other side. We report here the structure of a C-terminally truncated variant of human CK2α in which the enzyme adopts a decidedly inactive conformation for the first time. In this CK2α structure, those regulatory key regions still are in their active positions. Yet the glycine-rich ATP-binding loop, which is normally part of the canonical anti-parallel β-sheet, has collapsed into the ATP-binding site so that ATP is excluded from binding; specifically, the side chain of Arg47 occupies the ribose region of the ATP site and Tyr50, the space required by the triphospho moiety. We discuss some factors that may support or disfavor this inactive conformation, among them coordination of small molecules at a remote cavity at the CK2α/CK2β interaction region and binding of a CK2β dimer. The latter stabilizes the glycine-rich loop in the extended active conformation known from the majority of CK2α structures. Thus, the novel inactive conformation for the first time provides a structural basis for the stimulatory impact of CK2β on CK2α.
Co-reporter:Karsten Niefind, Christina W. Yde, Inessa Ermakova, Olaf-Georg Issinger
Journal of Molecular Biology (13 July 2007) Volume 370(Issue 3) pp:427-438
Publication Date(Web):13 July 2007
DOI:10.1016/j.jmb.2007.04.068
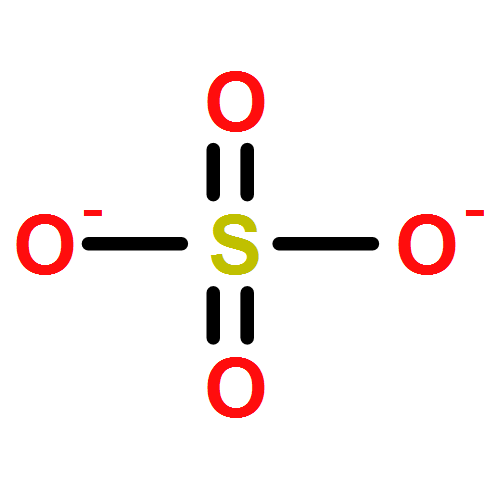
CK2α is the catalytic subunit of protein kinase CK2 and a member of the CMGC family of eukaryotic protein kinases like the cyclin-dependent kinases, the MAP kinases and glycogen-synthase kinase 3. We present here a 1.6 Å resolution crystal structure of a fully active C-terminal deletion mutant of human CK2α liganded by two sulfate ions, and we compare this structure systematically with representative structures of related CMGC kinases. The two sulfate anions occupy binding pockets at the activation segment and provide the structural basis of the acidic consensus sequence S/T-D/E-X-D/E that governs substrate recognition by CK2. The anion binding sites are conserved among those CMGC kinases. In most cases they are neutralized by phosphorylation of a neighbouring threonine or tyrosine side-chain, which triggers conformational changes for regulatory purposes. CK2α, however, lacks both phosphorylation sites at the activation segment and structural plasticity. Here the anion binding sites are functionally changed from regulation to substrate recognition. These findings underline the exceptional role of CK2α as a constitutively active enzyme within a family of strictly controlled protein kinases.
Co-reporter:Karsten Niefind, Christina W. Yde, Inessa Ermakova, Olaf-Georg Issinger
Journal of Molecular Biology (13 July 2007) Volume 370(Issue 3) pp:427-438
Publication Date(Web):13 July 2007
DOI:10.1016/j.jmb.2007.04.068
CK2α is the catalytic subunit of protein kinase CK2 and a member of the CMGC family of eukaryotic protein kinases like the cyclin-dependent kinases, the MAP kinases and glycogen-synthase kinase 3. We present here a 1.6 Å resolution crystal structure of a fully active C-terminal deletion mutant of human CK2α liganded by two sulfate ions, and we compare this structure systematically with representative structures of related CMGC kinases. The two sulfate anions occupy binding pockets at the activation segment and provide the structural basis of the acidic consensus sequence S/T-D/E-X-D/E that governs substrate recognition by CK2. The anion binding sites are conserved among those CMGC kinases. In most cases they are neutralized by phosphorylation of a neighbouring threonine or tyrosine side-chain, which triggers conformational changes for regulatory purposes. CK2α, however, lacks both phosphorylation sites at the activation segment and structural plasticity. Here the anion binding sites are functionally changed from regulation to substrate recognition. These findings underline the exceptional role of CK2α as a constitutively active enzyme within a family of strictly controlled protein kinases.



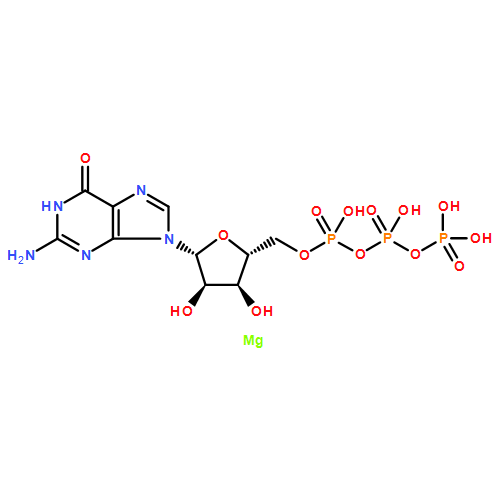






![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)