
Several decades of cumulated research evidence have proven that aggregation of beta-amyloid 42 (Aβ42) is the main cause of neuronal death in the brains of patients with Alzheimer's disease. Therefore, inhibition of Aβ42 aggregation holds great promise for the prevention and treatment of Alzheimer's disease. To this end, we used a systematic in vitro evolution including a paired peptide library method. We identified two peptides with high binding affinity (with Kd in the nm range) for Aβ42. Functionally, these peptides strongly inhibited the aggregation of Aβ42 as shown by the thioflavin T assay and atomic force microscopy. Moreover, these peptides rescued PC12 cells from the cytotoxic effect of aggregated Aβ42 in vitro. Our results suggest that these novel peptides may be potential therapeutic seeds for the treatment of Alzheimer's disease.
A method for efficient enrichment of protease inhibitors out of a DNA library was developed by introducing SF-link technology. A two-step selection strategy was designed consisting of the initial enrichment of aptamers based on binding function while the second enrichment step was based on the inhibitory activity to a protease, cathepsin E (CE). The latter was constructed by covalently linking of a biotinylated peptide substrate to each of the ssDNA molecule contained in the preliminarily selected DNA library, generating ‘SF-link’. Gradual enrichment of inhibitory DNAs was attained in the course of selection. One molecule, SFR-6-3, showed an IC50 of around 30 nM, a Kd of around 15 nM and high selectivity for CE. Sequence and structure analysis revealed a C-rich sequence without any guanine and possibly an i-motif structure, which must be novel to be found in in vitro-selected aptamers. SF-link technology, which is novel as the screening technology, provided a remarkable enrichment of specific protease inhibitors and has a potential to be further developed. Copyright © 2006 John Wiley & Sons, Ltd.









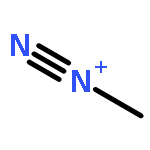



![1H-Imidazole-4-carboxamide,5-[2-(methylimino)hydrazinyl]-](http://img.cochemist.com/ccimg/3500/3413-72-7.png)
![1H-Imidazole-4-carboxamide,5-[2-(methylimino)hydrazinyl]-](http://img.cochemist.com/ccimg/3500/3413-72-7_b.png)








![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)