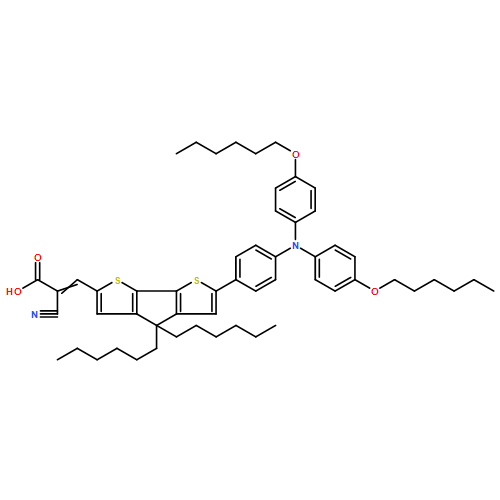
•Three novel dyes are prepared by tuning the steric hindrance of auxiliary donors.•Adsorption properties of dyes are studied with X-ray photoelectron spectroscopy.•Dye with largest auxiliary donor has the smallest tilt angle on TiO2.Three new D-D-π-A sensitizers ZHG5, ZHG6 and ZHG7 have been prepared by gradually improving the steric hindrance of auxiliary donors and the power conversion efficiencies (PCE) are 5.64%, 5.32% and 2.74%. UV–Vis absorption indicates that the molar extinction coefficients decrease with the increased steric hindrance of auxiliary donors. X-ray photoelectron spectroscopy (XPS) indicates that the tilt angles of ZHG5 and ZHG6 anchored on the TiO2 film are similar and ZHG7 is almost standing rather vertical on the TiO2 film with the smallest tilt angle. The results of dye desorption and XPS experiments indicate that the dye loading amount of ZHG6 with larger steric hindrance is lower than that of ZHG5, ZHG7 with largest auxiliary donor has the maximum loading amount probably due to its smallest tilt angle. Larger auxiliary donor of ZHG6 leads to higher open circuit voltage (Voc = 734 mV) but lower shorter circuit current (Jsc = 12.63 mA cm−2) compared with that of ZHG5 (Voc = 730 mV, Jsc = 12.06 mA cm−2). However, dense packing of dye ZHG7 anchored on the TiO2 leads to more serious intermolecular π-π aggregation effects. Perhaps this effect and lowest molar extinction coefficient are the reason that DSSC based on ZHG7 have the lowest PCE. So above results indicate that auxiliary donor with overlarge steric hindrance will have smaller tilt angle of dye anchored on TiO2 and may lead to more dye loading amount but serious intermolecular π-π aggregation effects.Auxiliary donor with overlarge steric hindrance will have smaller tilt angle of dye anchored on TiO2 and will lead to more dye loading amount but serious intermolecular π-π aggregation effects.Download high-res image (232KB)Download full-size image
Journal of Materials Science 2016 Volume 51( Issue 13) pp:6235-6248
Publication Date(Web):2016 July
DOI:10.1007/s10853-016-9921-8
We designed a series of 5,6,7-trithiapentacene-13-one (TTPO) derivatives (from 1 to 20) by attaching cyano groups (–CN) and by replacing S atoms with Se. The incoherent charge-hopping model combined with the Marcus–Hush electron transfer theory were used to evaluate the charge mobility of the title compounds as potential n-type organic field-effect transistors (OFETs) candidates. Based on the optimized face-to-face dimers, the crystal structures were predicted. The replacement of S with Se can decrease the electron reorganization energy (λ–). The introduction of −CN can decrease the energy levels of both HOMOs and LUMOs, which increases the stability. According to the electronic affinities (EA) and structures of dimers predicted by the DFT method coupled with dispersion corrections (−D), we selected compounds 2, 14, 15, and 19 as initial well-performance OFETs materials and predicted their crystal structures. Based on the optimized crystal structures, the charge (both electron and hole) transport mobilities are estimated. Electron mobilities of 14 and 15 are as high as 2.32 and 2.48 cm2 V−1 s−1, respectively, indicating that 14 and 15 are the promising n-type OFETs materials. What’s more, the predicted crystals show remarkable anisotropic charge mobilities. The maximum electron mobility appears along the direction of face-to-face π-stacking.
Co-reporter:Hai-Lang Jia, Ming-Dao Zhang, Ze-Min Ju, He-Gen Zheng and Xue-Hai Ju
Journal of Materials Chemistry A 2015 vol. 3(Issue 28) pp:14809-14816
Publication Date(Web):10 Jun 2015
DOI:10.1039/C5TA01857A
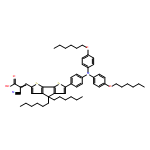
We developed a novel efficient tridentate anchoring group which can anchor dyes onto the TiO2 surface via synchronously choosing Lewis acid sites and Brønsted acid sites of TiO2. For the purpose of comparing the traditional carboxylate anchoring group to picolinic acid, two new D–π–A porphyrin dyes (JA1 and JA2) differing only in anchoring groups have been synthesized and applied in dye-sensitized solar cells. Picolinic acid as an anchoring group in the dye JA2 not only extended the scope of the spectral response, but also improved the charge transport properties and enhanced the electron injection efficiency. The PCE of the JA1 based-device (carboxylate as the anchoring group) was 5.76%. The PCE of the JA2 based-device was 7.20%, which increased by 25% compared with JA1. The dye TTR2 was used as a cosensitizer; it would not just make up for the poor absorption of porphyrin dyes in the 470–550 nm range, but also would suppress the main dye aggregation and reduce the charge recombination rate. We found that the picolinic acid anchor was more suitable for the cosensitization system than the carboxylate anchor, for there was almost no competitive adsorption between JA2 and TTR2. The JA2 + TTR2 based-device showed the highest PCE of 8.98% under AM 1.5 G irradiation.
The adsorption and dissociation mechanism of NH2NO2 on the Mg surface have been investigated by the generalized gradient approximation of density functional theory. Calculations employ a supercell (3 × 3 × 3) slab model and three-dimensional periodic boundary conditions. The strong attractive force between oxygen and Mg atoms induces the N–O bond of the NH2NO2 to decompose. The dissociated oxygen atoms and radical fragment of NH2NO2 oxidize readily Mg atoms. The largest adsorption energy is −860.5 kJ/mol. The largest charge transfer is 3.76 e from surface Mg atoms to fragments of NH2NO2. The energy barriers of N–O bond dissociation are in a range of 11.6–36.5 kJ/mol. The adsorption energy of NH2NO2 on the Mg surface compensates the energy needed for the N–O bond dissociation.
Co-reporter:Jian-Ying Zhao, Feng-Qi Zhao, Si-Yu Xu, and Xue-Hai Ju
The Journal of Physical Chemistry A 2013 Volume 117(Issue 10) pp:2213-2222
Publication Date(Web):February 20, 2013
DOI:10.1021/jp309422p
The adsorption and reaction of H2O molecule on neutral X-centered icosahedronal Al12X clusters (X = Al, Mg, Zn, Ga, Ni, Fe, B, C, Si, P) were investigated by PW91, PBE, and PWC methods. Reaction energies and reaction barriers were determined. The spin states and the doped atoms have important influences on the Al12X geometries, density, electronic properties, and energy density of reaction between Al12X with a single H2O molecule. The energies of the neutral X-centered Al12X are lower than that of surface X-replaced Al12X with the exception of Al12Mg. The H2O dissociation on the Al12X (X = Mg, Zn, Ga, Ni, Fe) clusters have relatively low activation barriers, but large activation barriers for Al12X (X = B, C, Si, P). The activation barrier of water dissociation on the singlet Al12Fe cluster is the lowest, whereas the highest barrier is with the Al12C. The reaction of H2O with Al12Fe is the most exothermic. The center-Fe atom can move out to the surface after the adsorption and dissociation of H2O with an energy barrier of 172 kJ/mol. The results showed that the water dissociation on the Al12X cluster can be tuned by controllable X doping.
The density functional theory method has been used to study the mechanism for the intramolecular hydrogen transfer (IHT) reaction of 1-methylbutly peroxide radical [n-C3H7CH(CH3)OO·]. The B3LYP method was used in conjunction with 6-31+G**, 6-311++G** and aug-cc-pVDZ basis sets. The geometrical configurations of reactants, productions and transition states were fully optimized on the potential energy surfaces. The results suggest that the IHT reactions involve the formation of transition states with five-, six- and seven-member rings when the transferring hydrogen atoms are at different initial positions related to the peroxide oxygen. The activation energies for the IHT reactions of β-, γ- and α-H migrations are 90.0, 100.8, and 140–152 kJ/mol at the B3LYP/6-311++G** level, respectively. The α-H is easier to transfer than the α- and γ-H. Both the activation energy and rate constant for β-H migrations at the B3LYP/6-31+G** level are basically in agreement with the experimental values. The methyl substituent on β-position slightly lowers the activation energy, which is consistent with the experimental fact. All the IHT reactions are endothermic. The β-H transferring is the most favorable process in view of both thermodynamics and kinetics. The predicted rate constants indicate that only the β-H and γ-H transfer reactions occur when the temperature is not high enough.
The adsorption of NH2NO2 molecule on the Al2O3(0 0 1) surface were investigated by the generalized gradient approximation (GGA) of density functional theory (DFT). The calculations employ a supercell model represented with 2 × 2 of periodic boundary conditions. The strong attractive forces between NH2NO2 molecule and Al2O3 induce obvious change of the NH2NO2 and Al2O3 structure. Although the NH2NO2 molecule does not decompose, AlO bonds partially decompose and some OO bonds form, whose bond lengths are intervenient between OO double bond length and OO single bond length. The largest adsorption energy is −453.8 kcal mol−1. By the adsorption energy and the change with structure of NH2NO2 and Al2O3, it can be concluded that aluminized explosive of NH2NO2 keeps high reactivity even if the aluminum is oxidized to form a film of the alumina. This finding can make clear the activated aluminum as a stored energy source for propellants and the good performance of aluminized explosives. The energies of DOS for N and O atoms of the NH2NO2 molecule match with those of Al atoms, and AlO or AlN bond forms easily at the corresponding energies range. The DOS projections on the N, O and Al atoms occur with obvious shift of peaks, which infers energy bands become broad and the interactions of chemical bonds are strengthened.
Chinese Journal of Chemistry 2012 Volume 30( Issue 10) pp:2539-2548
Publication Date(Web):
DOI:10.1002/cjoc.201200470
Abstract
The adsorption of 1,1-diamino-2,2-dinitroethylene (FOX-7) molecule on the Al(111) surface was investigated by the generalized gradient approximation (GGA) of density functional theory (DFT). The calculations employ a supercell (4×4×2) slab model and three-dimensional periodic boundary conditions. The strong attractive forces between oxygen and aluminum atoms induce the NO bond breaking of the FOX-7. Subsequently, the dissociated oxygen atoms and radical fragment of FOX-7 oxidize the Al surface. The largest adsorption energy is −940.5 kJ/mol. Most of charge transfer is 3.31e from the Al surface to the fragment of FOX-7 molecule. We also investigated the adsorption and decomposition mechanism of FOX-7 molecule on the Al(111) surface. The activation energy for the dissociation steps of P2 con?guration is as large as 428.8 kJ/mol, while activation energies of other con?gurations are much smaller, in range of 2.4 to 147.7 kJ/mol.
Journal of Structural Chemistry 2012 Volume 53( Issue 4) pp:659-664
Publication Date(Web):2012 July
DOI:10.1134/S0022476612040075
First-principle calculations are performed on the dimers of 2,6-diamino-3,5-dinitropyridine (ANPy) and its N-oxide (2,6-diamino-3,5-dinitropyridine-1-oxide, ANPyO). The dimers as well as the monomers are fully optimized by the DFT-B3LYP and HF methods in conjunction with 6-311G**, 6-311++G**, and cc-pVDZ basis sets. The N-O bond length of the pyridine N-oxide moiety decreases in the ANPyO dimer in the dimerization process, which results in a larger deformation energy of the ANPyO submolecule. This deformation prevents the submolecules from further close contact and the formation of strong H-bonds between the nitro and amino groups. The optimized intermolecular distances of the ANPyO dimer are in good agreement with the corresponding experimental values. There is a weak C-H...O hydrogen bond in the ANPyO dimer; the B3LYP method underestimates its binding energy. On the contrary, for the ANPy dimer, the binding energy obtained at the B3LYP level is larger than that obtained at the HF level. The individual O...H strength is stronger in the ANPy dimer than that in ANPyO, which is consistent with the O...H distance. The O...H-C type of the H-bond is stronger in the ANPyO dimer than the ordinary O...H-C bond due to the N-oxide oxygen atom bearing larger negative charges. The corrected binding energy for each hydrogen bond between nitro oxygen and amino hydrogen is about −5 kJ/mol in the ANPy dimer, which is stronger than that in the ANPyO dimer.
Journal of Cluster Science 2012 Volume 23( Issue 2) pp:395-410
Publication Date(Web):2012 June
DOI:10.1007/s10876-012-0441-7
Density functional theory method with full geometry optimization was used to study the adsorption of nitroamine (NH2NO2) on Al13 cluster. Both dissociative and nondissociative adsorption structures were predicted with different NH2NO2 molecule orientations on Al13 cluster surfaces. In dissociative chemisorption, the main decomposition products of NH2NO2 are O atom(s) and NH2NO or NH2N species. The O atoms being ruptured from the N–O bond form strong Al–O bonds with the neighboring Al around the adsorbed sites. In addition, the species obtained as a result of O atom elimination remains bonded to the surface. The largest adsorption energy is −737.66 kJ/mol when the NH2NO2 molecule decomposes into two O atoms and a NH2N fragment. For nondissociative adsorption, the seriously deformed nitroamine forms various N–O–Al bonding configurations with Al. The significant charge transfer occurs for all adsorption configurations. The most charge transfer is 2.068 e from the Al cluster surface to the fragments of the decomposed NH2NO2. The change of the electronic structures is obvious due to the adsorption or dissociation of NH2NO2 molecule. Nitroamine readily oxidizes the aluminum surface of the Al13 cluster.
Journal of Molecular Modeling 2011 Volume 17( Issue 2) pp:235-242
Publication Date(Web):2011 February
DOI:10.1007/s00894-010-0717-5
Geometrical structures and relative stabilities of (LiNH2)n (n = 1–5) clusters were studied using density functional theory (DFT) at the B3LYP/6-31G* and B3LYP/6-31++G* levels. The electronic structures, vibrational properties, N–H bond dissociation energies (BDE), thermodynamic properties, bond properties and ionization potentials were analyzed for the most stable isomers. The calculated results show that the Li–N and Li–Li bonds can be formed more easily than those of the Li–H or N–H bonds in the clusters, in which NH2 is bound to the framework of Li atomic clusters with fused rings. The average binding energies for each LiNH2 unit increase gradually from 142 kJ mol−1 up to about 180 kJ mol−1 with increasing n. Natural bond orbital (NBO) analysis suggests that the bonds between Li and NH2 are of strong ionicity. Three-center–two-electron Li–N–Li bonding exists in the (LiNH2)2 dimer. The N–H BDE values indicate that the change in N–H BDE values from the monomer a1 to the singlet-state clusters is small. The N–H bonds in singlet state clusters are stable, while the N–H bonds in triplet clusters dissociate easily. A study of their thermodynamic properties suggests that monomer a1 forms clusters (b1, c1, d2 and e1) easily at low temperature, and clusters with fewer numbers of rings tend to transfer to ones with more rings at low temperature. Eg, EHOMO and Eav decrease gradually, and become constant. Ring-like (LiNH2)3,4 clusters possess higher ionization energy (VIE) and Eg, but lower values of EHOMO. Ring-like (LiNH2)3,4 clusters are more stable than other types. A comparison of structures and spectra between clusters and crystal showed that the NH2 moiety in clusters has a structure and spectral features similar to those of the crystal.
Co-reporter:Xiaowei Fan;Chenggang Gu;Gong Chen; Dr. Xuehai Ju
Chinese Journal of Chemistry 2010 Volume 28( Issue 12) pp:2364-2370
Publication Date(Web):
DOI:10.1002/cjoc.201190005
Abstract
Density functional calculations at the B3LYP level with 6-311G** and aug-cc-pVDZ basis sets were performed to predict the heats of formation (HOFs) for two pyrazine derivatives and eight pyridine derivatives. In the isodesmic reactions designed for the computation of heats of formation (HOFs), pyrazine and pyridine were chosen as reference compounds. The N-oxidations for the ring nitrogen of pyrazine and pyridine derivatives decrease the HOF values when N-oxide oxygen is neighboring with amino groups, but increase when it neighbors with nitro groups. Thermal stability was evaluated via bond dissociation energies (BDE) at the UB3LYP/6-311G** level. As a whole, the homolysis of C–NO2 bonds is the main step for bond dissociation of the title compounds. The BDE values of title compounds are influenced by intramolecular hydrogen bonds. The hydrogen bond effects associated with the length of the H···O bonds were analyzed by the electron density at the critical points and natural bond orbital.
Co-reporter:Su-Qin Zhou, Feng-Qi Zhao, Xue-Hai Ju, Xiao-Chun Cheng and Jian-Hua Yi
The Journal of Physical Chemistry C 2010 Volume 114(Issue 20) pp:9390-9397
Publication Date(Web):May 5, 2010
DOI:10.1021/jp101137c
The density functional theory generalized gradient approximation has been used to study the adsorption of nitroamine molecules on the Al(111) surface. The calculations employ a 4 × 4 aluminum slab with three layers and three-dimensional periodic boundary conditions. There exist both physical and chemical adsorptions associated with different NH2NO2 molecule orientations and particular aluminum surface sites. For the nondissociative adsorption, the nitro oxygen atom orients to the Al surface. In the case of dissociative chemisorption, the O and N atoms bind with the Al surface. The O and N atoms of broken down N−O and N−N bonds form strong Al−O and Al−N bonds with the neighboring Al sites around the dissociation sites. Moreover, the radical species obtained as a result of N−O and N−N bond dissociation remains bonded to the surface. The largest adsorption energy is −893.8 kJ/mol. For the dissociation adsorption configurations, a significant charge transfer occurs. The most charge transfer is 3.04 e from the Al surface to the NH2NO2 molecule. The change of the electronic structures is obvious due to the dissociation of the N−O and N−N bonds and the formation of strong Al−O and Al−N bonds. It can be inferred that the aluminum surface is readily oxidized by the adsorbate of nitroamine, by dissociation of either the O and N atoms from the nitro group or the N atom from the amino group.
Homodesmotic reactions were designed for the computation of strain energies (SE) for four nitro-substituted 1,3,5,7-tetraazacubane derivatives. Total energies of the optimized geometric structures at the DFT-B3LYP/6-31G* and DFT-B3LYP/6-311G** levels were used to derive the SE. The variation of SE with respect to the number of substituents is similar with both basis sets. The SE value is 237.32 kcal/mol at the B3LYP/6-311G** level for 2,4,6,8-tetranitro-1,3,5,7-tetraazacubane, which is unexpectedly much larger than that of its cubane analogue. The SE increases remarkably with more nitro groups being attached to the cage skeleton of tetraazacubane. The ‘bending’ of the bonds within the cubic skeleton attributes to the increase of strains as the attached number of nitro groups increases.
Co-reporter:Yu-Fang Li, Zun-Yao Wang, Xue-Hai Ju, Xiao-Wei Fan
Journal of Molecular Structure: THEOCHEM 2009 Volume 907(1–3) pp:29-34
Publication Date(Web):15 August 2009
DOI:10.1016/j.theochem.2009.04.012
Density functional theory (DFT) calculations were performed for a series of 2,2′-bi-1H-imidazole derivatives. The B3LYP and B3P86 functionals with 6-311G∗∗ basis set were used. The heats of formation (HOFs) were predicted through designed isodesmic reactions. Calculated results show that the HOFs decrease as the –NO2 groups being replaced by –NF2, but the HOFs increase as the –NO2 groups being replaced by –N3. When the –NO2 groups are replaced by –NH2, the HOFs initially decrease then increase. As for the isomeric compounds, the HOFs decrease following the increase of the number of hydrogen bonds for the different substituent position. At B3LYP/6-311G∗∗ level, the HOF of 4,4′-diamino-5,5′-dinitro-2,2′-bi-1H-imidazole is the smallest (125.2 kJ/mol), and the HOF of 4,4′,5,5′-tetraazido-2,2′-bi-1H-imidazole is the largest (1608.9 kJ/mol). The magnitudes of intramolecular group interactions were predicted through the disproportionation energies. The Edisproportion of 4,4′-diamino-5,5′-dinitro-2,2′-bi-1H-imidazole is −70.3 kJ/mol and is the smallest among the title compounds, but that of 4,4′,5,5′-tetranitro-2,2′-bi-1H-imidazole is the largest, which is 128.1 kJ/mol. Thermal stabilities were evaluated via bond dissociation energies (BDE) at the UB3LYP/6-311G∗∗ level. The BDEZPE value for C–NO2 bond, 270.0 kJ/mol in average, is the smallest compared to other types of bonds.
Co-reporter:Yu-Fang Li, Xiao-Wei Fan, Zun-Yao Wang, Xue-Hai Ju
Journal of Molecular Structure: THEOCHEM 2009 Volume 896(1–3) pp:96-102
Publication Date(Web):28 February 2009
DOI:10.1016/j.theochem.2008.11.004
Computations by density functional theory (DFT) method were performed on a series of pyrazole derivatives. The heats of formation (HOFs) were predicted using B3LYP and B3P86 functionals with aug-cc-pVDZ and 6-311++G∗∗ basis sets via designed isodesmic reactions. In the isodesmic reactions the pyrazole was chosen as a reference compound. The general trend is that the HOFs increase with increasing number of –NH2 and –N3 groups. And the HOFs initially decrease then increase as the number –NO2 and –NF2 groups increasing. The HOF of 3,5-didifluoroaminopyrazole is the smallest (134.4 kJ/mol), and the HOF of 3,4,5-triazidopyrazole is the largest (1240.6 kJ/mol) at the B3LYP/aug-cc-pVDZ level. The position of the group also affects the HOFs. Judged by the HOF values, the 4- and 3-monosubstituted isomers are the most stable monosubstituted isomers when the electron pulling and pushing groups are attached to the pyrazole ring, respectively. The 3,5-isomers are the most stable di-substituted isomers. The values of HOFs decrease as the –NO2 groups being replaced by the –NH2 and –NF2, but increase dramatically as the –NO2 groups being replaced by –N3 with the average increment of 332.0 kJ/mol. The relative stability of the title compounds was evaluated based on the calculated HOFs and the energy gaps between the frontier orbits. Thermal stabilities were evaluated via bond dissociation energies (BDE) at the UB3LYP/aug-cc-pVDZ level. The value of C–N3 bond dissociation energy, 524.5 kJ/mol in average, is relatively larger than other out-of-ring C–N bonds. The BDEZPE value of C–NO2 bond is smaller than all the other C–N bonds, which is 270.7 kJ/mol in average. These results provide basic information for the molecular design of novel high energetic density materials.
Co-reporter:Hui-Ming Zhao, Xiao-Yan Wei, Zun-Yao Wang, Xue-Hai Ju
Journal of Molecular Structure: THEOCHEM 2007 Volume 817(1–3) pp:5-10
Publication Date(Web):1 September 2007
DOI:10.1016/j.theochem.2007.04.021
Heats of formation (HOFs) and strengths of group interactions for 19 polyisocyanoadamantanes were obtained by using the density functional theory (DFT). The adamantane skeleton was chosen for a reference compound in the process of designing isodesmic reactions. It was found that the HOF increases 213–252 kJ/mol for each additional number of the isocyano group being added to the adamantane skeleton. The HOFs slightly deviate from the group additivity with respect to the isocyano group. The distance between isocyano groups influences the values of HOFs. The disproportionation energies of neighbor isocyano groups in polyisocyanoadamantanes are in the range of 10.3–15.8 kJ/mol, which are also slightly related to the substituent numbers. The average interaction energy between nearest neighbor –NC group in decaisocyanoadamantane is 10.34 kJ/mol at the B3LYP/6-311G∗∗ level. The relative stability related to the number of isocyano groups of the title compounds was evaluated based on the calculated HOFs, the energy gaps between the frontier orbitals, the bond orders of the C–NC and C–C bonds, and bond dissociation energies.
Journal of Molecular Structure: THEOCHEM 2007 Volume 804(1–3) pp:95-100
Publication Date(Web):16 March 2007
DOI:10.1016/j.theochem.2006.10.017
The hybrid DFT methods with 12 different basis sets were used for the computation of FOOF. Comparison with CCSD/6-311 + G* method was made. The availability and shortcoming of DFT methods for FOOF were pointed out. On that basis, the binary compounds of fluorine and oxygen (OnFm, n = 1–3, m = 1–2) were calculated at the G96PW91/D95(3df) level. The affiliation of lone pair electrons toward the fluorine atom decreases from OF2 to O3F2 as the number of oxygen increases. The strength of O–O bond decreases from O2F2 to O3F2. The shorter O–F bond of O3F2 could readily change to longer one and vice versa through a transition state with energy barrier of 11.06 kJ/mol. The O–F bond is much stronger than the O–O bond in O3F2. On the contrary, the O–F bond is much weaker than the O–O bond in O2F2. O2F and O3F can be regarded as weakly bound OO–F and OO–OF adducts, respectively. O3F2 could be regarded as a weakly bound FO–OOF adduct. The standard enthalpies and free energies of formation for the binary compounds were predicted at both the G96PW91 and the G2 levels.
Both the heats of formation (HOFs) and the group interactions were predicted for a series of adamantane derivatives substituted with difluoroamino groups by using density functional theory (DFT) with 6-31G∗ and 6-311G∗∗ basis sets. The adamantane skeleton was chosen for as a reference compound in the isodesmic reactions designed for calculating the HOFs. The contribution of difluoroamino group to the heat of formation deviates from group additivity. The relationship between HOFs and molecular structures was discussed. It was found that the HOFs decrease more with each difluoroamino group attached to the tertiary carbon than to the secondary carbon of the adamantane skeleton, when the secondary carbon was substituted by one group only. However, the HOFs increase with the second difluoroamino group attached to the secondary carbon. The distance between difluoroamino groups influences the values of HOFs. The group interacting energies of polydifluoroaminoadamantane increase in the range of 8–70 kJ/mol with each difluoroamino group attached. The interaction of neighbor difluoroamino groups discords with the group additivity. The relative stability of the title compounds was assessed based on the calculated HOFs, the energy gaps between the frontier orbitals, and the bond order of CNF2. The CNF2 bond is greatly weakened with two NF2 groups being attached to the same secondary carbon. The basis sets influence the HOFs when the substituent number is over 6 or when two substituted groups being attached to the same secondary carbon.
Co-reporter:Hai-Lang Jia, Ming-Dao Zhang, Ze-Min Ju, He-Gen Zheng and Xue-Hai Ju
Journal of Materials Chemistry A 2015 - vol. 3(Issue 28) pp:NaN14816-14816
Publication Date(Web):2015/06/10
DOI:10.1039/C5TA01857A
We developed a novel efficient tridentate anchoring group which can anchor dyes onto the TiO2 surface via synchronously choosing Lewis acid sites and Brønsted acid sites of TiO2. For the purpose of comparing the traditional carboxylate anchoring group to picolinic acid, two new D–π–A porphyrin dyes (JA1 and JA2) differing only in anchoring groups have been synthesized and applied in dye-sensitized solar cells. Picolinic acid as an anchoring group in the dye JA2 not only extended the scope of the spectral response, but also improved the charge transport properties and enhanced the electron injection efficiency. The PCE of the JA1 based-device (carboxylate as the anchoring group) was 5.76%. The PCE of the JA2 based-device was 7.20%, which increased by 25% compared with JA1. The dye TTR2 was used as a cosensitizer; it would not just make up for the poor absorption of porphyrin dyes in the 470–550 nm range, but also would suppress the main dye aggregation and reduce the charge recombination rate. We found that the picolinic acid anchor was more suitable for the cosensitization system than the carboxylate anchor, for there was almost no competitive adsorption between JA2 and TTR2. The JA2 + TTR2 based-device showed the highest PCE of 8.98% under AM 1.5 G irradiation.

![2,6-Diphenyldithieno[3,2-b:2',3'-d]thiophene](http://img.cochemist.com/ccimg/881900/881838-94-4.png)
![2,6-Diphenyldithieno[3,2-b:2',3'-d]thiophene](http://img.cochemist.com/ccimg/881900/881838-94-4_b.png)
![DITHIENO[3,2-B:2',3'-D]THIOPHENE, 2,6-BIS(PENTAFLUOROPHENYL)-](http://img.cochemist.com/ccimg/864000/863913-58-0.png)
![DITHIENO[3,2-B:2',3'-D]THIOPHENE, 2,6-BIS(PENTAFLUOROPHENYL)-](http://img.cochemist.com/ccimg/864000/863913-58-0_b.png)
![1,3,4,6-TETRANITRO-2,3A,5,6A-TETRAHYDROIMIDAZO[4,5-D]IMIDAZOLE](http://img.cochemist.com/ccimg/473800/473796-67-7.png)
![1,3,4,6-TETRANITRO-2,3A,5,6A-TETRAHYDROIMIDAZO[4,5-D]IMIDAZOLE](http://img.cochemist.com/ccimg/473800/473796-67-7_b.png)










![Benzo[1,2-b:3,4-b':5,6-b'']tris[1]benzothiophene](http://img.cochemist.com/ccimg/200/199-14-4.png)
![Benzo[1,2-b:3,4-b':5,6-b'']tris[1]benzothiophene](http://img.cochemist.com/ccimg/200/199-14-4_b.png)

