Co-reporter:Kuljeet Kaur;Sergio Parra;Rong Chen
Naunyn-Schmiedeberg's Archives of Pharmacology 2012 Volume 385( Issue 5) pp:443-453
Publication Date(Web):2012 May
DOI:10.1007/s00210-011-0705-z
Receptors coupled to G proteins have many effects on the heart. Enhanced signaling by Gαs and Gαq leads to cardiac injury and heart failure, while Gαi2 signaling in cardiac myocytes can protect against ischemic injury and β-adrenergic-induced heart failure. We asked whether enhanced Gαi2 signaling in mice could protect against heart failure using a point mutation in Gαi2 (G184S), which prevents negative regulation by regulators of G protein signaling. Contrary to our expectation, it worsened effects of a genetic dilated cardiomyopathy (DCM) and catecholamine-induced cardiac injury. Gαi2G184S/+/DCM double heterozygote mice (TG9+Gαi2G184S/+) had substantially decreased survival compared to DCM animals. Furthermore, heart weight/body weight ratios (HW/BW) were significantly greater in TG9+Gαi2G184S/+ mice as was expression of natriuretic peptide genes. Catecholamine injury in Gαi2G184S/G184S mutant mice produced markedly increased isoproterenol-induced fibrosis and collagen III gene expression vs WT mice. Cardiac fibroblasts from Gαi2G184S/G184S mice also showed a serum-dependent increase in proliferation and ERK phosphorylation, which were blocked by pertussis toxin and a mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor. Gαi2 signaling in cardiac myocytes protects against ischemic injury but enhancing Gαi2 signaling overall may have detrimental effects in heart failure, perhaps through actions on cardiac fibroblasts.
Co-reporter:Levi L. Blazer, Haoming Zhang, Emma M. Casey, Stephen M. Husbands, and Richard R. Neubig
Biochemistry 2011 Volume 50(Issue 15) pp:
Publication Date(Web):February 17, 2011
DOI:10.1021/bi1019622
Regulators of G-protein signaling (RGS) proteins are potent negative modulators of signal transduction through G-protein-coupled receptors. They function by binding to activated (GTP-bound) Gα subunits and accelerating the rate of GTP hydrolysis. Modulation of RGS activity by small molecules is an attractive mechanism for fine-tuning GPCR signaling for therapeutic and research purposes. Here we describe the pharmacologic properties and mechanism of action of CCG-50014, the most potent small molecule RGS inhibitor to date. It has an IC50 for RGS4 of 30 nM and is >20-fold selective for RGS4 over other RGS proteins. CCG-50014 binds covalently to the RGS, forming an adduct on two cysteine residues located in an allosteric regulatory site. It is not a general cysteine alkylator as it does not inhibit activity of the cysteine protease papain at concentrations >3000-fold higher than those required to inhibit RGS4 function. It is also >1000-fold more potent as an RGS4 inhibitor than are the cysteine alkylators N-ethylmaleimide and iodoacetamide. Analysis of the cysteine reactivity of the compound shows that compound binding to Cys107 in RGS8 inhibits Gα binding in a manner that can be reversed by cleavage of the compound−RGS disulfide bond. If the compound reacts with Cys160 in RGS8, the adduct induces RGS denaturation, and activity cannot be restored by removal of the compound. The high potency and good selectivity of CCG-50014 make it a useful tool for studying the functional roles of RGS4.
Co-reporter:Chris R. Evelyn, Jessica L. Bell, Jenny G. Ryu, Susan M. Wade, Andrew Kocab, Nicole L. Harzdorf, H.D. Hollis Showalter, Richard R. Neubig, Scott D. Larsen
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 2) pp:665-672
Publication Date(Web):15 January 2010
DOI:10.1016/j.bmcl.2009.11.056
We recently identified bis(amide) CCG-1423 (1) as a novel inhibitor of RhoA/C-mediated gene transcription that is capable of inhibiting invasion of PC-3 prostate cancer cells in a Matrigel model of metastasis. An initial structure–activity relationship study focusing on bioisosteric replacement of the amides and conformational restriction identified two compounds, 4g and 8, with improved selectivity for inhibition of RhoA/C-mediated gene transcription and attenuated cytotoxicity relative to 1. Both compounds were also capable of inhibiting cell invasion with equal efficacy to 1 but with less attendant cytotoxicity.Conformational restriction and bioisosteric amide replacement were used to generate novel analogs of the Rho-transcriptional pathway inhibitor CCG-1423. Two compounds were found with significantly improved selectivity for inhibition of PC-3 cancer cell invasion versus cytotoxicity.
Co-reporter:Levi L Blazer and Richard R Neubig
Neuropsychopharmacology 2009 34(1) pp:126-141
Publication Date(Web):September 17, 2008
DOI:10.1038/npp.2008.151
Protein–protein interactions are a crucial element in cellular function. The wealth of information currently available on intracellular-signaling pathways has led many to appreciate the untapped pool of potential drug targets that reside downstream of the commonly targeted receptors. Over the last two decades, there has been significant interest in developing therapeutics and chemical probes that inhibit specific protein–protein interactions. Although it has been a challenge to develop small molecules that are capable of occluding the large, often relatively featureless protein–protein interaction interface, there are increasing numbers of examples of small molecules that function in this manner with reasonable potency. This article will highlight the current progress in the development of small molecule protein–protein interaction inhibitors that have applications in the treatment or study of central nervous system function and disease. In particular, we will focus upon recent work towards developing small molecule inhibitors of amyloid-β and α-synuclein aggregation, inhibitors of critical components of G-protein-signaling pathways, and PDZ domain inhibitors.
Co-reporter:Rebecca A Roof;David L Roman;Samuel T Clements
BMC Pharmacology 2009 Volume 9( Issue 1) pp:
Publication Date(Web):2009 December
DOI:10.1186/1471-2210-9-9
Regulators of G protein signaling (RGSs) accelerate GTP hydrolysis by Gα subunits and profoundly inhibit signaling by G protein-coupled receptors (GPCRs). The distinct expression patterns and pathophysiologic regulation of RGS proteins suggest that inhibitors may have therapeutic potential. We recently described a focused one-bead, one-compound (OBOC) library screen to identify peptide inhibitors of RGS4. Here we extend our observations to include another peptide with a different mechanism of action.Peptide 5nd (Tyr-Trp-c [Cys-Lys-Gly-Leu-Cys]-Lys-NH2, S-S) blocks the RGS4-Gαo interaction with an IC50 of 28 μM. It forms a covalent, dithiothreitol (DTT) sensitive adduct with a mass consistent with the incorporation of one peptide per RGS. Peptide 5nd activity is abolished by either changing its disulfide bridge to a methylene dithioether bridge, which cannot form disulfide bridges to the RGS, or by removing all cysteines from the RGS protein. However, no single cysteine in RGS4 is completely necessary or sufficient for 5nd activity.Though it has some RGS selectivity, 5nd appears to be a partially random cysteine modifier. These data suggest that it inhibits RGS4 by forming disulfide bridges with the protein.
Co-reporter:Donald J. Bernsteel, David L. Roman, Richard R. Neubig
Analytical Biochemistry 2008 Volume 383(Issue 2) pp:180-185
Publication Date(Web):15 December 2008
DOI:10.1016/j.ab.2008.08.026
Protein kinases are important drug targets, and a wide variety of methods have been developed for assessing their activity. A key element in developing selective kinase inhibitors is the ability to rapidly compare the effects of an inhibitor on several related or unrelated kinases. We describe a simple, nonradioactive, bead-based method for detecting kinase activity in vitro. Biotinylated peptide substrates are immobilized on beads and phosphorylation is detected with anti-phosphopeptide antibodies with no separation steps required. Phosphorylation is dependent on the amount of kinase in the assay and can be inhibited by known kinase inhibitors in a concentration-dependent manner. Using Luminex technology, we measured the activity of three kinases (PKA, PKC-μ, and Akt) on multiple substrates simultaneously. We also discuss conditions necessary to optimize measurement of the activity of several kinases in a single sample.





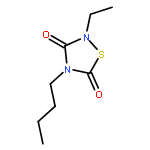
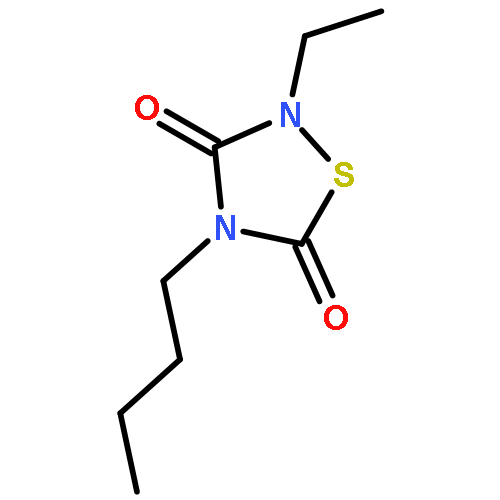
















![3H-Pyrrolo[4,3,2-gh]-1,4-benzodiazonin-3-one,1,2,4,5,6,8-hexahydro-5-(hydroxymethyl)-1-methyl-2-(1-methylethyl)-, (2S,5S)-](http://img.cochemist.com/ccimg/90400/90365-57-4.png)
![3H-Pyrrolo[4,3,2-gh]-1,4-benzodiazonin-3-one,1,2,4,5,6,8-hexahydro-5-(hydroxymethyl)-1-methyl-2-(1-methylethyl)-, (2S,5S)-](http://img.cochemist.com/ccimg/90400/90365-57-4_b.png)