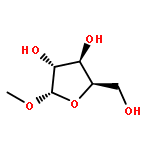
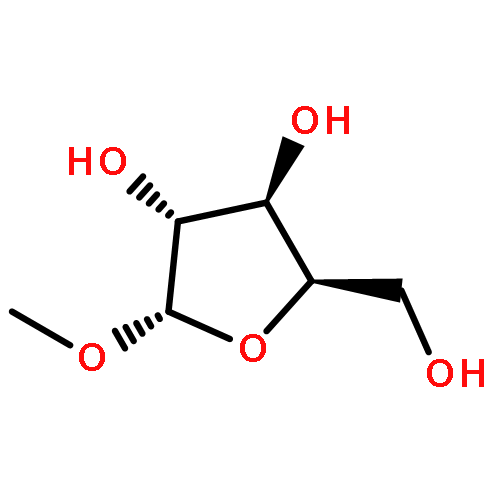
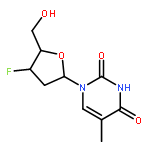
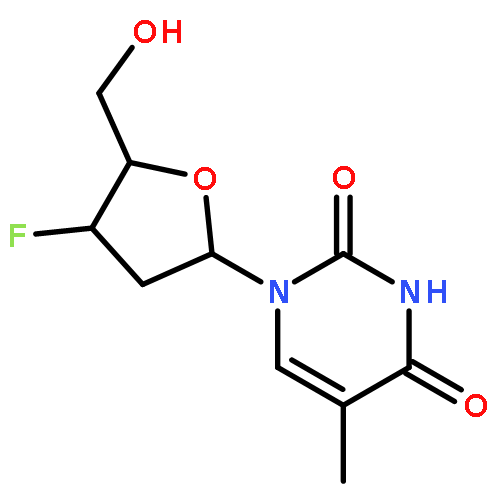
Co-reporter:Karam Ch;Rakesh K Tiwari;Sumit Kumar;Amir Nasrolahi Shirazi;Sweta Sharma;Erik V Van der Eycken;Virinder S Parmar;Sunil K Sharma
Journal of Heterocyclic Chemistry 2015 Volume 52( Issue 2) pp:562-572
Publication Date(Web):
DOI:10.1002/jhet.2106
A new class of 4-oxo-4H-1-benzopyran derivatives were synthesized and their antiproliferative activity examined against a panel of three human cancer cell lines, that is, breast carcinoma (MDA-MB-468), ovarian adenocarcinoma (SK-OV-3), and colorectal adenocarcinoma (HT-29). Two compounds, that is, 3-hexyl-7,8-dihydroxy-4-oxo-4H-1-benzopyran and (E)-ethyl 3-(7-methoxy-4-oxo-4H-1-benzopyran-3-yl)acrylate were found to be potent against all three cancer cell lines studied at 50 μM concentration. Also, the inhibitory potency of the compounds was evaluated against active Src kinase. A few of these compounds exhibited modest Src kinase inhibitory activity (IC50 = 52–57 μM). Structure-activity relationship studies with respect to the nature and position of substituents on the lead compounds could be further exploited for the design and development of more potent antiproliferative agents and/or Src kinase inhibitors.
Co-reporter:Bhanu Pemmaraju, Hitesh K. Agarwal, Donghoon Oh, Karen W. Buckheit, Robert W. Buckheit Jr., Rakesh Tiwari, Keykavous Parang
Tetrahedron Letters 2014 Volume 55(Issue 12) pp:1983-1986
Publication Date(Web):19 March 2014
DOI:10.1016/j.tetlet.2014.02.001
A number of 5′-O-dicarboxylic fatty acyl monoester derivatives of 3′-azido-3′-deoxythymidine (zidovudine, AZT), 2′,3′-didehydro-2′,3′-dideoxythymidine (stavudine, d4T), and 3′-fluoro-3′-deoxythymidine (alovudine, FLT) were synthesized to improve the lipophilicity and potentially the cellular delivery of parent polar 2′,3′-dideoxynucleoside (ddN) analogs. The compounds were evaluated for their anti-HIV activity. Three different fatty acids with varying chain length of suberic acid (octanedioic acid), sebacic acid (decanedioic acid), and dodecanedioic acid were used for the conjugation with the nucleosides. The compounds were evaluated for anti-HIV activity and cytotoxicity. All dicarboxylic ester conjugates of nucleosides exhibited significantly higher anti-HIV activity than that of the corresponding parent nucleoside analogs. Among all the tested conjugates, 5′-O-suberate derivative of AZT (EC50 = 0.10 nM) was found to be the most potent compound and showed 80-fold higher anti-HIV activity than AZT without any significant toxicity (TC50 >500 nM).
Co-reporter:Naglaa Salem El-Sayed, Amir Nasrolahi Shirazi, Magda Goda El-Meligy, Ahmed Kamel El-Ziaty, David Rowley, Jiadong Sun, Zenat Adeeb Nagib, Keykavous Parang
Tetrahedron Letters 2014 Volume 55(Issue 6) pp:1154-1158
Publication Date(Web):5 February 2014
DOI:10.1016/j.tetlet.2013.12.081
A novel class of 6-indolypyridine-3-carbonitrile derivatives were synthesized and evaluated for antiproliferative activities to establish structure–activity relationship. The synthesis was carried out through one-pot multicomponent reaction of 3-acetylindole, aromatic aldehydes, ethyl cyanoacetate, and ammonium acetate in the presence of piperidine as a catalyst, using a microwave irradiation method or a traditional thermal method. This was followed by chlorination for compounds 13a–e and subsequent nucleophilic substitution of the chlorine group by ethylenediamine at C2 position of the pyridine ring. The antiproliferative activity of these new nicotinonitriles was evaluated against human ovarian adenocarcinoma (SK-OV-3), breast adenocarcinoma (MCF-7), and cervix adenocarcinoma (HeLa) cells. Among all compounds, 2-((2-aminoethyl)amino)-4-aryl-6-indolylnicotinonitriles series (15a, 15b, 15d, and 15e) exhibited higher antiproliferative activity on the three cancer cell lines with IC50 values of 4.1–13.4 μM.
Co-reporter:Somayeh Motavallizadeh, Asal Fallah-Tafti, Saeedeh Maleki, Amir Nasrolahi Shirazi, Mahboobeh Pordeli, Maliheh Safavi, Sussan Kabudanian Ardestani, Shaaban Asd, Rakesh Tiwari, Donghoon Oh, Abbas Shafiee, Alireza Foroumadi, Keykavous Parang, Tahmineh Akbarzadeh
Tetrahedron Letters 2014 Volume 55(Issue 2) pp:373-375
Publication Date(Web):8 January 2014
DOI:10.1016/j.tetlet.2013.11.033
Several novel N-(9-oxo-9H-xanthen-4-yl)benzenesulfonamide derivatives were prepared as potential antiproliferative agents. The in vitro antiproliferative activity of the synthesized compounds was investigated against a panel of tumor cell lines including breast cancer cell lines (MDA-MB-231, T-47D) and neuroblastoma cell line (SK-N-MC) using MTT colorimetric assay. Etoposide, a well-known anticancer drug, was used as a positive standard drug. Among synthesized compounds, 4-methoxy-N-(9-oxo-9H-xanthen-4-yl)benzenesulfonamide (5i) showed the highest antiproliferative activity against MDA-MB-231, T-47D, and SK-N-MC cells. Furthermore, pentafluoro derivatives 5a and 6a exhibited higher antiproliferative activity than doxorubicin against human leukemia cell line (CCRF-CEM) and breast adenocarcinoma (MDA-MB-468) cells. Structure–activity relationship studies revealed that xanthone benzenesulfonamide hybrid compounds can be used for the development of new lead anticancer agents.
Co-reporter:Donghoon Oh;Dr. Shaban Anwar Darwish;Dr. Amir Nasrolahi Shirazi; Rakesh Kumar Tiwari; Keykavous Parang
ChemMedChem 2014 Volume 9( Issue 11) pp:2449-2453
Publication Date(Web):
DOI:10.1002/cmdc.201402230
Abstract
Two bicyclic peptides composed of tryptophan and arginine residues were synthesized from monocyclic peptide building blocks and evaluated as cellular delivery agents. [W5G]-(triazole)-[KR5] and [W5E]-(β-Ala)-[KR5] containing triazole and β-alanine linkers improved the cellular delivery of fluorescein (F′)-labeled phosphopeptide F′-GpYEEI (F′-PP) by 7.6- and 19.3-fold, respectively, in human ovarian adenocarcinoma cells. However, parent monocyclic peptide [R5] and monocyclic peptide [WR]4 only enhanced the cellular uptake of the phosphopeptide by only 1.3- and 3.7-fold, respectively. Confocal microscopy showed that the corresponding fluorescein-labeled bicyclic peptide F′-[KW4E]-(β-Ala)-[KR5] was localized in the cytosol and nucleus. Studying the cellular uptake of F′-[KW4E]-(β-Ala)-[KR5] in the presence of endocytosis inhibitors indicated that the clathrin- and caveolin-dependent endocytosis are the main pathways for cellular uptake. The bicyclic peptide was able to improve antiproliferative activity of doxorubicin by 20 %. These data suggest that this bicyclic peptide can be utilized as a new class of cell-penetrating peptides and cellular delivery tools.
Co-reporter:Karam Chand, Suchita Prasad, Rakesh K. Tiwari, Amir N. Shirazi, Sumit Kumar, Keykavous Parang, Sunil K. Sharma
Bioorganic Chemistry 2014 53() pp: 75-82
Publication Date(Web):
DOI:10.1016/j.bioorg.2014.02.001
Co-reporter:V. Kameshwara Rao, Ganesh M. Shelke, Rakesh Tiwari, Keykavous Parang, and Anil Kumar
Organic Letters 2013 Volume 15(Issue 9) pp:2190-2193
Publication Date(Web):April 15, 2013
DOI:10.1021/ol400738r
An efficient and simple strategy has been developed for the synthesis of 2,3-diarylnaphthofurans using sequential hydroarylation of naphthols and alkynes in the presence of In(OTf)3 under microwave irradiation followed by one-pot Heck-oxyarylation of generated 1-substituted-α-hydroxy styrenes.
Co-reporter:Hitesh K. Agarwal, Bhupender S. Chhikara, Sitaram Bhavaraju, Dindyal Mandal, Gustavo F. Doncel, and Keykavous Parang
Molecular Pharmaceutics 2013 Volume 10(Issue 2) pp:467-476
Publication Date(Web):August 23, 2012
DOI:10.1021/mp300361a
Three fatty acyl conjugates of (−)-2′,3′-dideoxy-5-fluoro-3′-thiacytidine (FTC, emtricitabine) were synthesized and evaluated against HIV-1 cell-free and cell-associated virus and compared with the corresponding parent nucleoside and physical mixtures of FTC and fatty acids. Among all the compounds, the myristoylated conjugate of FTC (5, EC50 = 0.07–3.7 μM) displayed the highest potency. Compound 5 exhibited 10–24 and 3–13-times higher anti-HIV activity than FTC alone (EC50 = 0.7–88.6 μM) and the corresponding physical mixtures of FTC and myristic acid (14, EC50 = 0.2–20 μM), respectively. Cellular uptake studies confirmed that compound 5 accumulated intracellularly after 1 h of incubation and underwent intracellular hydrolysis in CCRF-CEM cells. Alternative studies were conducted using the carboxyfluorescein conjugated with FTC though β-alanine (12) and 12-aminododecanoic acid (13). Acylation of FTC with a long-chain fatty acid in 13 improved its cellular uptake by 8.5–20 fold in comparison to 12 with a short-chain β-alanine. Compound 5 (IC90 = 15.7–16.1 nM) showed 6.6- and 35.2 times higher activity than FTC (IC90 = 103–567 nM) against multidrug resistant viruses B-NNRTI and B–K65R, indicating that FTC conjugation with myristic acid generates a more potent analogue with a better resistance profile than its parent compound.Keywords: (−)-2′,3′-dideoxy-5-fluoro-3′-thiacytidine; anti-HIV; cellular uptake; cytotoxicity; fatty acids;
Co-reporter:Amir Nasrolahi Shirazi, Rakesh Tiwari, Bhupender S. Chhikara, Dindyal Mandal, and Keykavous Parang
Molecular Pharmaceutics 2013 Volume 10(Issue 2) pp:488-499
Publication Date(Web):January 9, 2013
DOI:10.1021/mp3004034
Doxorubicin (Dox) is a hydrophilic anticancer drug that has short retention time due to the efficient efflux in some cancer cells (e.g., ovarian adenocarcinoma SK-OV-3). Cyclic [W(RW)4] and the corresponding linear peptide (RW)4 were conjugated with Dox through an appropriate linker to afford cyclic [W(RW)4]–Dox and linear (RW)4–Dox conjugates to enhance the cellular uptake and cellular retention of the parent drug for sustained anticancer activity. Comparative antiproliferative assays between covalent (cyclic [W(RW)4]–Dox and linear (RW)4–Dox) and the corresponding noncovalent physical mixtures of the peptides and Dox were performed. Cyclic [W(RW)4]–Dox inhibited the cell proliferation of human leukemia (CCRF-CEM) (62–73%), ovarian adenocarcinoma (SK-OV-3) (51–74%), colorectal carcinoma (HCT-116) (50–67%), and breast carcinoma (MDA-MB-468) (60–79%) cells at a concentration of 1 μM after 72–120 h of incubation. Cyclic [W(RW)4]–Dox exhibited higher antiproliferative activity than linear (RW)4–Dox in all cancer cells with the highest activity observed after 72 h. Flow cytometry analysis showed 3.6-fold higher cellular uptake of cyclic [W(RW)4]–Dox than Dox alone in SK-OV-3 cells after 24 h incubation. The cellular hydrolysis study showed that 99% of cyclic [W(RW)4]–Dox was hydrolyzed intracellularly within 72 h and released Dox. These data suggest that cyclic [W(RW)4]–Dox can be used as a potential prodrug for improving the cellular delivery and retention of Dox.Keywords: anticancer; cell-penetrating peptides; cellular uptake; cyclic peptides; doxorubicin;
Co-reporter:Dindyal Mandal, Rakesh K. Tiwari, Amir Nasrolahi Shirazi, Donghoon Oh, Guofeng Ye, Antara Banerjee, Arpita Yadav and Keykavous Parang
Soft Matter 2013 vol. 9(Issue 39) pp:9465-9475
Publication Date(Web):07 Aug 2013
DOI:10.1039/C3SM50764E
A number of cyclic peptides including [FR]4, [FK]4, [WK]5, [CR]4, [AK]4, and [WR]n (n = 3–5) containing L-amino acids were produced using solid-phase peptide synthesis. We hypothesized that an optimal balance of hydrophobicity and charge could generate self-assembled nanostructures in aqueous solution by intramolecular and/or intermolecular interactions. Among all the designed peptides, [WR]n (n = 3–5) generated self-assembled vesicle-like nanostructures at room temperature as shown by transmission electron microscopy (TEM), scanning electron microscopy (SEM), and/or dynamic light scattering (DLS). This class of peptides represents the first report of surfactant-like cyclic peptides that self-assemble into nanostructures. A plausible mechanistic insight into the self-assembly of [WR]5 was obtained by molecular modeling studies. Modified [WR]5 analogues, such as [WMeR]5, [WR(Me)2]5, [WMeR(Me)2]5, and [WdR]5, exhibited different morphologies to [WR]5 as shown by TEM observations. [WR]5 exhibited a significant stabilizing effect for generated silver nanoparticles and glyceraldehyde-3-phosphate dehydrogenase activity. These studies established a new class of surfactant-like cyclic peptides that self-assembled into nanostructures and could have potential applications for the stabilization of silver nanoparticles and protein biomolecules.
Co-reporter:Amir Nasrolahi Shirazi, Rakesh Kumar Tiwari, Donghoon Oh, Antara Banerjee, Arpita Yadav, and Keykavous Parang
Molecular Pharmaceutics 2013 Volume 10(Issue 5) pp:2008-2020
Publication Date(Web):March 28, 2013
DOI:10.1021/mp400046u
Phosphopeptides are valuable reagent probes for studying protein–protein and protein–ligand interactions. The cellular delivery of phosphopeptides is challenging because of the presence of the negatively charged phosphate group. The cellular uptake of a number of fluorescent-labeled phosphopeptides, including F′-GpYLPQTV, F′-NEpYTARQ, F′-AEEEIYGEFEAKKKK, F′-PEpYLGLD, F′-pYVNVQN-NH2, and F′-GpYEEI (F′ = fluorescein), was evaluated in the presence or absence of a [WR]4, a cyclic peptide containing alternative arginine (R) and tryptophan (W) residues, in human leukemia cells (CCRF-CEM) after 2 h incubation using flow cytometry. [WR]4 improved significantly the cellular uptake of all phosphopeptides. PEpYLGLD is a sequence that mimics the pTyr1246 of ErbB2 that is responsible for binding to the Chk SH2 domain. The cellular uptake of F′-PEpYLGLD was enhanced dramatically by 27-fold in the presence of [WR]4 and was found to be time-dependent. Confocal microscopy of a mixture of F′-PEpYLGLD and [WR]4 in live cells exhibited intracellular localization and significantly higher cellular uptake compared to that of F′-PEpYLGLD alone. Transmission electron microscopy (TEM) and isothermal calorimetry (ITC) were used to study the interaction of PEpYLGLD and [WR]4. TEM results showed that the mixture of PEpYLGLD and [WR]4 formed noncircular nanosized structures with width and height of 125 and 60 nm, respectively. ITC binding studies confirmed the interaction between [WR]4 and PEpYLGLD. The binding isotherm curves, derived from sequential binding models, showed an exothermic interaction driven by entropy. These studies suggest that amphiphilic peptide [WR]4 can be used as a cellular delivery tool of cell-impermeable negatively charged phosphopeptides.Keywords: cellular uptake; cyclic peptides; nanoparticles; phosphopeptides; SH2 domain;
Co-reporter:Amir Nasrolahi Shirazi, Donghoon Oh, Rakesh Kumar Tiwari, Brian Sullivan, Anju Gupta, Geoffrey D. Bothun, and Keykavous Parang
Molecular Pharmaceutics 2013 Volume 10(Issue 12) pp:4717-4727
Publication Date(Web):October 29, 2013
DOI:10.1021/mp400539r
Peptide amphiphiles (PAs) are promising tools for the intracellular delivery of numerous drugs. PAs are known to be biodegradable systems. Here, four PA derivatives containing arginine and lysine conjugated with fatty acyl groups with different chain lengths, namely, PA1: R-K(C14)-R, PA2: R-K(C16)-R, PA3: K(C14)-R-K(C14), and PA4: K(C16)-R-K(C16), where C16 = palmitic acid and C14 = myristic acid, were synthesized through Fmoc chemistry. Flow cytometry studies showed that, among all synthesized PAs, only K(C16)-R-K(C16), PA4 was able to enhance the cellular uptake of a fluorescence-labeled anti-HIV drug 2′,3′-dideoxy-3′-thiacythidine (F′-3TC, F′ = fluorescein) and a biologically important phosphopeptide (F′-PEpYLGLD) in human leukemia cells (CCRF-CEM) after 2 h incubation. For example, the cellular uptake of F′-3TC and F′-PEpYLGLD was enhanced approximately 7.1- and 12.6-fold in the presence of the PA4 compared to those of the drugs alone. Confocal microscopy of F′-3TC and F′-PEpYLGLD loaded PA4 in live cells showed significantly higher intracellular localization than the drug alone in human ovarian cells (SK-OV-3) after 2 h incubation. The high-performance liquid chromatography (HPLC) results showed that loading of Dox by the peptide amphiphile was 56% after 24 h. The loaded Dox was released (34%) within 48 h intracellularly. The circular dichrosim (CD) results exhibited that the secondary structure of the peptide was changed upon interactions with Dox. Mechanistic studies revealed that endocytosis is the major pathway of the internalization. These studies suggest that PAs containing the appropriate sequence of amino acids, chain length, charge, and hydrophobicity can be used as cellular delivery tools for transporting drugs and biomolecules.Keywords: cellular uptake; drug delivery; nuclear targeting; peptide amphiphiles;
Co-reporter:Amir Nasrolahi Shirazi, Rakesh Kumar Tiwari, Donghoon Oh, Brian Sullivan, Kellen McCaffrey, Dindyal Mandal, and Keykavous Parang
Molecular Pharmaceutics 2013 Volume 10(Issue 8) pp:3137-3151
Publication Date(Web):July 8, 2013
DOI:10.1021/mp400199e
Gold nanoparticles (AuNPs) were synthesized in situ in a green and rapid method from the reaction of reducing linear and cyclic peptides containing tryptophan and lysine residues, (KW)5 and cyclic [KW]5, with an aqueous solution of HAuCl4 and were evaluated as cellular nanodrug delivery systems. The cyclic or linear nature of the peptide was found to determine the morphology and size of the formed peptide-AuNPs and their in vitro molecular transporting efficiency. While cyclic [KW]5-AuNPs formed sponge-like agglomerates, linear (KW)5-AuNPs demonstrated ball-shaped structures. A comparative flow cytometry study showed that the cellular uptake of fluorescence-labeled anti-HIV drugs (emtricitabine (FTC) and lamivudine (3TC)) in human leukemia (CCRF-CEM) cells, and a negatively charged cell-impermeable phosphopeptide (GpYEEI) in human ovarian adecarcinoma (SK-OV-3) cells was significantly higher in the presence of cyclic [KW]5-AuNPs than that of linear (KW)5-AuNPs, parent cyclic [KW]5, and linear (KW)5 peptides. For example, the cellular uptake of F′-GpYEEI was enhanced 12.8-fold by c[KW]5-AuNPs. Confocal microscopy revealed the localization of fluorescence-labeled-3TC in the presence of c[KW]5-AuNPs mostly in nucleus in SK-OV-3 cells after 1 h. On the other hand, l(KW)5-AuNPs delivered fluorescence-labeled-3TC in cytoplasm. These data suggest that noncell penetrating peptides can be converted to efficient molecular transporters through peptide-capped AuNPs formation.Keywords: cellular uptake; cyclic peptide; drug delivery; gold nanoparticles; lysine; nuclear targeting; tryptophan;
Co-reporter:Amir Nasrolahi Shirazi, Dindyal Mandal, Rakesh K. Tiwari, Liangran Guo, Wei Lu, and Keykavous Parang
Molecular Pharmaceutics 2013 Volume 10(Issue 2) pp:500-511
Publication Date(Web):September 21, 2012
DOI:10.1021/mp300448k
Co-reporter:Amir Nasrolahi Shirazi, Rakesh Kumar Tiwari, Alex Brown, Dindyal Mandal, Gongqin Sun, Keykavous Parang
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 11) pp:3230-3234
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmcl.2013.03.124
A number of cyclic and linear peptides containing various combinations of amino acids were evaluated for their Src kinase inhibitory potency. Among all the peptides, cyclic decapeptide C[RW]5 containing alternative arginine (R) and tryptophan (W) residues was found to be the most potent Src kinase inhibitor. C[RW]5 showed higher inhibitory activity (IC50 = 2.8 μM) than C[KW]5, L(KW)5, C[RW]4, and C[RW]3 with IC50 values of 46.9, 69.1, 21.5, and 25.0 μM, respectively, as determined in a fluorescence intensity-based assay. Thus, the cyclic nature, the presence of arginine, ring size, and the number of amino acids in the structure of the peptide were found to be critical in Src kinase inhibitory potency. The IC50 value of C[RW]5 was found to be 0.8 μM in a radioactive assay using [γ-32P]-ATP and polyE4Y as the substrate. C[RW]5 was a noncompetitive Src kinase inhibitor, showing approximately fourfold more selectivity towards Src than Abl.Cyclic peptides containing tryptophan and arginine are reported as a new class of Src kinase inhibitors.
Co-reporter:Kasiviswanadharaju Pericherla, Amir Nasrolahi Shirazi, V. Kameshwara Rao, Rakesh K. Tiwari, Nicholas DaSilva, Kellen T. McCaffrey, Yousef A. Beni, Antonio González-Sarrías, Navindra P. Seeram, Keykavous Parang, Anil Kumar
Bioorganic & Medicinal Chemistry Letters 2013 23(19) pp: 5329-5331
Publication Date(Web):
DOI:10.1016/j.bmcl.2013.07.058
Co-reporter:Hitesh K. Agarwal ; Bhupender S. Chhikara ; Michael J. Hanley ; Guofeng Ye ; Gustavo F. Doncel
Journal of Medicinal Chemistry 2012 Volume 55(Issue 10) pp:4861-4871
Publication Date(Web):April 25, 2012
DOI:10.1021/jm300492q
A number of fatty acyl derivatives of (−)-2′,3′-dideoxy-3′-thiacytidine (lamivudine, 3TC, 1) were synthesized and evaluated for their anti-HIV activity. The monosubstituted 5′-O-fatty acyl derivatives of 3TC (EC50 = 0.2–2.3 μM) were more potent than the corresponding monosubstituted N4-fatty acyl (EC50 = 0.4–29.4 μM) and 5′-O-N4-disubstituted (EC50 = 72.6 to >154.0 μM) derivatives of the nucleoside. 5′-O-Myristoyl (16) and 5′-O-12-azidododecanoyl derivatives (17) were found to be the most potent compounds (EC50 = 0.2–0.9 μM) exhibiting at least 16–36-fold higher anti-HIV activity against cell-free virus than 1 (EC50 = 11.4–32.7 μM). The EC90 values for 16 against B-subtype and C-subtype clinical isolates were several folds lower than those of 1. The cellular uptake studies confirmed that compound 16 accumulated intracellularly after 1 h of incubation with CCRF-CEM cells and underwent intracellular hydrolysis. 5′-O-Fatty acyl derivatives of 1 showed significantly higher anti-HIV activity than the corresponding physical mixtures against the B-subtype virus.
Co-reporter:Bhupender S. Chhikara ; Deendayal Mandal
Journal of Medicinal Chemistry 2012 Volume 55(Issue 4) pp:1500-1510
Publication Date(Web):January 25, 2012
DOI:10.1021/jm201653u
A number of lipophilic 14-substituted derivatives of doxorubicin were synthesized through conjugation of doxorubicin-14-hemisuccinate with different fatty amines or tetradecanol to enhance the lipophilicity, cellular uptake, and cellular retention for sustained anticancer activity. The conjugates inhibited the cell proliferation of human leukemia (CCRF-CEM, 69–76%), colon adenocarcinoma (HT-29, 60–77%), and breast adenocarcinoma (MDA-MB-361, 66–71%) cells at a concentration of 1 μM after 96–120 h of incubation. The N-tetradecylamido derivative of doxorubicin 14-succinate (10) exhibited consistently comparable antiproliferative activity to doxorubicin in a time-dependent manner (IC50 = 77 nM in CCRF-CEM cells). Flow cytometry analysis showed a 3-fold more cellular uptake of 10 than doxorubicin in SK-OV-3 cells. Confocal microscopy revealed that the conjugate was distributed in cytoplasmic and perinuclear areas during the first 1 h of incubation and slowly relocalized in the nucleus after 24 h. The cellular hydrolysis study showed that 98% of compound 10 was hydrolyzed intracellularly within 48 h and released doxorubicin.
Co-reporter:Hitesh K. Agarwal ; Bhupender S. Chhikara ; Megrose Quiterio ; Gustavo F. Doncel
Journal of Medicinal Chemistry 2012 Volume 55(Issue 6) pp:2672-2687
Publication Date(Web):February 21, 2012
DOI:10.1021/jm201551m
Mono-, di-, and trinucleoside conjugates of glutamate or peptide scaffolds containing nucleoside reverse transcriptase inhibitors were synthesized. Among dinucleoside glutamate ester derivatives, N-myristoylated derivatives showed significantly higher anti-HIV activity than the corresponding N-acetylated conjugates against cell-free virus. Myristoyl-Glu(3TC)-FLT (46, EC50 = 0.3–0.6 μM) and myristoyl-Glu(FTC)-FLT (47, EC50 = 0.1–0.4 μM) derivatives were the most active glutamate–dinucleoside conjugates. A trinucleoside glutamate derivative containing AZT, FLT, and 3TC (34, EC50 = 0.9–1.4 μM) exhibited higher anti-HIV activity than AZT and 3TC against cell-free virus. Compound 34 also exhibited higher anti-HIV activity against multidrug (IC50 = 5.9 nM) and NNRTI (IC50 = 12.9 nM) resistant viruses than parent nucleosides. The physical mixture containing FLT–succinate, AZT, 3TC, and glutamic acid exhibited 115-fold less activity against cell associated virus (EC50 = 91.9 μM) when compared to 34 (EC50 = 0.8 μM). Other conjugates showed less or comparable potency to that of the corresponding physical mixtures.
Co-reporter:Manoj Kumar Muthayala, Bhupender S. Chhikara, Keykavous Parang, and Anil Kumar
ACS Combinatorial Science 2012 Volume 14(Issue 1) pp:60
Publication Date(Web):October 20, 2011
DOI:10.1021/co200149m
An ionic liquid-supported aldehyde was designed and converted to ionic liquid-supported secondary aryl amines through reductive amination. The reaction of ionic liquid-supported aryl amines with sulfonyl chlorides and acid chlorides, respectively, followed by cleavage using trifluoroacetic acid (TFA) afforded sulfonamides and caboxamides. To introduce additional diversity in the synthesis of sulfonamides and caboxamides, ionic liquid-supported iodosubstituted aryl amine was synthesized using the same strategy, and underwent Suzuki coupling reaction, followed by reaction with a methanesulfonyl chloride to generate the corresponding biaryl sulfonamide. The advantages of the protocol over solid-phase synthesis are homogeneous reaction medium, high loading, easy separation of products, and characterization of intermediates.Keywords: carboxamides; ionic liquids; parallel synthesis; sulfonamides; Suzuki coupling reaction
Co-reporter:Raman K. Sharma, Sukhdev Singh, Rakesh Tiwari, Deendayal Mandal, Carl E. Olsen, Virinder S. Parmar, Keykavous Parang, Ashok K. Prasad
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 23) pp:6821-6830
Publication Date(Web):1 December 2012
DOI:10.1016/j.bmc.2012.09.057
A series of peracetylated O-aryl α,β-d-ribofuranosides have been synthesized and an efficient biocatalytic methodology has been developed for the separation of their anomers which was otherwise almost impossible by column chromatographic or other techniques. The incubation of 2,3,5-tri-O-acetyl-1-O-aryl-α,β-d-ribofuranoside with Lipozyme® TL IM immobilized on silica led to the selective deacetylation of only one acetoxy group, viz the C-5′-O-acetoxy group of the α-anomer over the other acetoxy groups derived from the two secondary hydroxyl groups present in the molecule and also over three acetoxy groups (derived from one primary and two secondary hydroxyls of the β-anomer). This methodology led to the easy synthesis of both, α- and β-anomers of O-aryl d-ribofuranosides. All the arylribofuranosides were screened for inhibition of Src kinase. 1-O-(3-Methoxyphenyl)-β-d-ribofuranoside exhibited the highest activity for inhibition of Src kinase (IC50 = 95.0 μM).
Co-reporter:V. Kameshwara Rao, Bhupender S. Chhikara, Rakesh Tiwari, Amir Nasrolahi Shirazi, Keykavous Parang, Anil Kumar
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 1) pp:410-414
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmcl.2011.10.124
A number of 2-substituted tetrahydroindazolones were synthesized by three-component condensation reaction of 1,3-diketones, substituted hydrazines, benzaldehydes, and Yb(OTf)3 as a catalyst in [bmim][BF4] ionic liquid using a simple, efficient, and economical one-pot method. The synthesized tetrahydroindazolones were evaluated for inhibition of cell proliferation of human colon carcinoma (HT-29), human ovarian adenocarcinoma (SK-OV-3), and c-Src kinase activity. 3,4-Dichlorophenyl tetrahydroindazolone derivative (15) inhibited the cell proliferation of HT-29 and SK-OV-3 cells by 62% and 58%, respectively. 2,3-Diphenylsubstituted tetrahydroindazolone derivatives, 19, 25, and 33, inhibited the cell proliferation of HT-29 cells by 65–72% at a concentration of 50 μM. In general, the tetrahydroindazolones showed modest inhibition of c-Src kinase where 4-tertbutylphenyl- (32) and 3,4-dichlorophenyl- (13) derivatives showed the inhibition of c-Src kinase with IC50 values of 35.1 and 50.7 μM, respectively.One-pot synthesis, Src kinase inhibitory potencies, and anticancer activities of 2-substituted tetrahydroindazolones are reported.
Co-reporter:Hitesh K. Agarwal, Karen W. Buckheit, Robert W. Buckheit Jr., Keykavous Parang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 17) pp:5451-5454
Publication Date(Web):1 September 2012
DOI:10.1016/j.bmcl.2012.07.037
Three nucleoside analogues, 3′-fluoro-2′,3′-dideoxythymidine (FLT), 3′-azido-2′,3′-dideoxythymidine (AZT), and 2′,3′-dideoxy-3′-thiacytidine (3TC) were conjugated with three different dicarboxylic acids to afford the long chain dicarboxylate esters of nucleosides. In general, dinucleoside ester conjugates of FLT and 3TC with long chain dicarboxylic acids exhibited higher anti-HIV activity than their parent nucleosides. Dodecanoate and tetradecanoate dinucleoside ester derivatives of FLT were found to be the most potent compounds with EC50 values of 0.8–1.0 nM and 3–4 nM against HIV-1US/92/727 and HIV-1IIIB cells, respectively. The anti-HIV activity of the 3TC conjugates containing long chain dicarboxylate diester (EC50 = 3–60 nM) was improved by 1.5–66 fold when compared to 3TC (EC50 = 90–200 nM). This study reveals that the symmetrical ester conjugation of dicarboxylic acids with a number of nucleosides results in conjugates with improved anti-HIV profile.The synthesis and anti-HIV activities of symmetrical dicarboxylate esters of dinucleoside reverse transcriptase inhibitors are reported.
Co-reporter:Anil Kumar, Manoj Kumar Muthyala, Sunita Choudhary, Rakesh K. Tiwari, and Keykavous Parang
The Journal of Organic Chemistry 2012 Volume 77(Issue 20) pp:9391-9396
Publication Date(Web):September 30, 2012
DOI:10.1021/jo301607a
A convenient synthesis of 1,2,3-thiadiazoles and 1,2,3-selenadiazoles was achieved using an ionic liquid as a novel soluble support. Ionic liquid-supported sulfonyl hydrazine was synthesized and reacted with a number of ketones to afford the corresponding ionic liquid-supported hydrazones that were converted to 1,2,3-thiadiazoles in the presence of thionyl chloride. The reaction of ionic liquid-supported hydrazones with selenium dioxide in acetonitrile afforded 1,2,3-selenadiazoles. The advantages of this methodology were the ease of workup, simple reaction conditions, and high purity.
Co-reporter:Asal Fallah-Tafti, Alireza Foroumadi, Rakesh Tiwari, Amir Nasrolahi Shirazi, David G. Hangauer, Yahao Bu, Tahmineh Akbarzadeh, Keykavous Parang, Abbas Shafiee
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 10) pp:4853-4858
Publication Date(Web):October 2011
DOI:10.1016/j.ejmech.2011.07.050
KX2-391 (KX-01/Kinex Pharmaceuticals), N-benzyl-2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetamide, is a highly selective Src substrate binding site inhibitor. To understand better the role of pyridine ring and N-benzylsubstitution in KX2-391 and establish the structure–activity relationship, a number of N-benzyl substituted (((2-morpholinoethoxy)phenyl)thiazol-4-yl)acetamide derivatives containing thiazole instead of pyridine were synthesized and evaluated for Src kinase inhibitory activities. The unsubstituted N-benzyl derivative (8a) showed the inhibition of c-Src kinase with GI50 values of 1.34 μM and 2.30 μM in NIH3T3/c-Src527F and SYF/c-Src527F cells, respectively. All the synthesized compounds were evaluated for inhibition of cell proliferation of human colon carcinoma (HT-29), breast carcinoma (BT-20), and leukemia (CCRF-CEM) cells. 4-Fluorobenzylthiazolyl derivative 8b exhibited 64–71% inhibition in the cell proliferation of BT-20 and CCRF cells at concentration of 50 μM.A number of N-benzyl substituted (2-morpholinoethoxy)phenyl)thiazol-4-yl)acetamide derivatives were synthesized and their Src kinase inhibitory and anti-proliferation activities of the conjugates were evaluated and compared in leukemia, breast, and colon cancer cell lines.Highlights► A number of N-benzyl-substituted acetamide derivatives were synthesized. ► The compounds were evaluated as Src kinase inhibitors and anticancer agents. ► The unsubstituted N-benzyl derivative was the most potent Src kinase inhibitor. ► 4-Fluorobenzylthiazolyl derivative inhibited the proliferation of breast cancer.
Co-reporter:Bhupender S. Chhikara, Nicole St. Jean, Deendayal Mandal, Anil Kumar, Keykavous Parang
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 6) pp:2037-2042
Publication Date(Web):June 2011
DOI:10.1016/j.ejmech.2011.02.056
Doxorubicin is extensively used in anticancer therapy. Doxorubicin is highly hydrophilic, has short half-life, and its use is associated with severe side effects at high doses. Fatty acyl amide derivatives of doxorubicin were synthesized with the expectation to improve the lipophilicity and anticancer activity of the drug. The lipophilicity was enhanced with the increase in chain length of fatty acyl moiety. Conjugation of 4′-amino group with fatty acids through an amide bond reduced the anticancer activity in leukemia, breast, ovarian, and colon cancer cell lines, suggesting that the presence of free amino group is required for anticancer activity of doxorubicin. Dodecanoyl-doxorubicin derivative was consistently the most effective among the synthesized derivatives and inhibited the proliferation of colon (HT-29) and ovarian (SK-OV-3) cancer cells by 64% and 58%, respectively, at a concentration of 1 μM after 96 h incubation.A number of lipophilic derivatives of doxorubicin were synthesized and their antiproliferation activities of the conjugates were evaluated and compared in leukemia, breast, ovarian, and colon cancer cell lines.Highlights► Fatty acyl amide derivatives of doxorubicin were synthesized and evaluated. ► Dodecanoyl derivative was the most effective among the synthesized derivatives. ► Dodecanoyl derivative inhibited the proliferation of colon and ovarian cancer cells. ► Free amino group is required for anticancer activity of doxorubicin.
Co-reporter:Dalip Kumar, V. Buchi Reddy, Anil Kumar, Deendayal Mandal, Rakesh Tiwari, Keykavous Parang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 1) pp:449-452
Publication Date(Web):1 January 2011
DOI:10.1016/j.bmcl.2010.10.121
Two classes of 1,4-disubstituted 1,2,3-triazoles were synthesized using one-pot reaction of α-tosyloxy ketones/α-halo ketones, sodium azide, and terminal alkynes in the presence of aq PEG (1:1, v/v) using the click chemistry approach and evaluated for Src kinase inhibitory activity. Structure–activity relationship analysis demonstrated that insertion of C6H5– and 4-CH3C6H4– at position 4 for both classes and less bulkier aromatic group at position 1 in class 1 contribute critically to the modest Src inhibition activity (IC50 = 32–43 μM) of 1,4-disubstituted 1,2,3-triazoles.The synthesis and Src kinase inhibitory activity of two classes of 1,4-disubstituted 1,2,3-triazoles are reported.
Co-reporter:Chandravanu Dash, Yousef Ahmadibeni, Michael J. Hanley, Jui Pandhare, Mathias Gotte, Stuart F.J. Le Grice, Keykavous Parang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 12) pp:3519-3522
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmcl.2011.05.005
Despite the success of potent reverse transcriptase (RT) inhibitors against human immunodeficiency virus type 1 (HIV-1) in combination regimens, the development of drug resistant RTs constitutes a major hurdle for the long-term efficacy of current antiretroviral therapy. Nucleoside β-triphosphate analogs of adenosine and nucleoside reverse transcriptase inhibitors (NRTIs) (3′-azido-2′,3′-dideoxythymidine (AZT), 3′-fluoro-2′,3′-dideoxythymidine (FLT), and 2′,3′-didehydro-2′,3′-dideoxythymidine (d4T)) were synthesized and their inhibitory activities were evaluated against wild-type and multidrug resistant HIV-1 RTs. Adenosine β-triphosphate (1) and AZT β-triphosphate (2) completely inhibited the DNA polymerase activity of wild type, the NRTI multi resistant, and nonnucleoside RT inhibitors (NNRTI) resistant HIV-1 RT at 10 nM, 10 and 100 μM, respectively.The synthesis and inhibitory activities of nucleoside β-triphosphate analogs of adenosine, zidovudine, alovudine, and stavudine against wild-type and multidrug resistant HIV-1 reverse transcriptases are reported.
Co-reporter:V. Kameshwara Rao, Bhupender S. Chhikara, Amir Nasrolahi Shirazi, Rakesh Tiwari, Keykavous Parang, Anil Kumar
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 12) pp:3511-3514
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmcl.2011.05.010
An efficient and economical method was developed for the synthesis of 3-substituted indoles by one-pot three-component coupling reaction of a substituted or unsubstituted benzaldehyde, N-methylaniline, and indole or N-methylindole using Yb(OTf)3–SiO2 as a catalyst. All the synthesized compounds were evaluated for inhibition of cell proliferation of human colon carcinoma (HT-29), human ovarian adenocarcinoma (SK-OV-3), and c-Src kinase activity. The 4-methylphenyl (4o and 4p) and 4-methoxyphenyl (4q) indole derivatives inhibited the cell proliferation of SK-OV-3 and HT-29 cells by 70–77% at a concentration of 50 μM. The unsubstituted phenyl (4d) and 3-nitrophenyl (4l) derivatives showed the inhibition of c-Src kinase with IC50 values of 50.6 and 58.3 μM, respectively.One-pot synthesis, Src kinase inhibitory potencies, and anticancer activities of 3-substituted indoles are reported.
Co-reporter:Hitesh K. Agarwal, Kelly Loethan, Deendayal Mandal, Gustavo F. Doncel, Keykavous Parang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 7) pp:1917-1921
Publication Date(Web):1 April 2011
DOI:10.1016/j.bmcl.2011.02.070
A number of 5′-O-fatty acyl derivatives of 2′,3′-didehydro-2′,3′-dideoxythymidine (stavudine, d4T) were synthesized and evaluated for anti-HIV activities against cell-free and cell-associated virus, cellular cytotoxicity, and cellular uptake studies. The conjugates were found to be more potent than d4T. Among these conjugates, 5′-O-12-azidododecanoyl derivative of d4T (2), displaying EC50 = 3.1–22.4 μM, showed 4- to 9-fold higher activities than d4T against cell-free and cell-associated virus. Cellular uptake studies were conducted on CCRF-CEM cell line using 5(6)-carboxyfluorescein derivatives of d4T attached through β-alanine (9) or 12-aminododecanoic acid (10) as linkers. The fluorescein-substituted analog of d4T with long chain length (10) showed 12- to 15-fold higher cellular uptake profile than the corresponding analog with short chain length (9). These studies reveal that conjugation of fatty acids to d4T enhances the cellular uptake and anti-HIV activity of stavudine.The synthesis, anti-HIV activities against cell-free and cell-associated virus, cellular cytotoxicity, and cellular uptake studies of 5′-O-fatty acyl derivatives of 2′,3′-didehydro-2′,3′-dideoxythymidine (stavudine, d4T) are reported.
Co-reporter:Anil Kumar, Israr Ahmad, Bhupender S. Chhikara, Rakesh Tiwari, Deendayal Mandal, Keykavous Parang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 5) pp:1342-1346
Publication Date(Web):1 March 2011
DOI:10.1016/j.bmcl.2011.01.047
A series of two classes of 3-phenylpyrazolopyrimidine-1,2,3-triazole conjugates were synthesized using click chemistry approach. All compounds were evaluated for inhibition of Src kinase and human ovarian adenocarcinoma (SK-Ov-3), breast carcinoma (MDA-MB-361), and colon adenocarcinoma (HT-29). Hexyl triazolyl-substituted 3-phenylpyrazolopyrimidine exhibited inhibition of Src kinase with an IC50 value of 5.6 μM. 4-Methoxyphenyl triazolyl-substituted 3-phenylpyrazolopyrimidine inhibited the cell proliferation of HT-29 and SK-Ov-3 by 73% and 58%, respectively, at a concentration of 50 μM.The synthesis, Src kinase inhibitory potencies, and anticancer activities of two classes of 3-phenylpyrazolopyrimidine-1,2,3-triazole conjugates are reported.
Co-reporter:Dr. Deendayal Mal;Amir NasrolahiShirazi ; Keykavous Parang
Angewandte Chemie International Edition 2011 Volume 50( Issue 41) pp:9633-9637
Publication Date(Web):
DOI:10.1002/anie.201102572
Co-reporter:Dr. Deendayal Mal;Amir NasrolahiShirazi ; Keykavous Parang
Angewandte Chemie 2011 Volume 123( Issue 41) pp:9807-9811
Publication Date(Web):
DOI:10.1002/ange.201102572
Co-reporter:Y. Ahmadibeni, C. Dash, M. J. Hanley, S. F. J. Le Grice, H. K. Agarwal and K. Parang
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 6) pp:1271-1274
Publication Date(Web):13 Jan 2010
DOI:10.1039/B922846B
A polymer-bound α,β-methylene-β-triphosphitylating reagent was synthesized and subjected to reactions with unprotected nucleosides, followed by oxidation, deprotection of cyanoethoxy groups, and acidic cleavage to afford nucleoside 5′-O-α,β-methylene-β-triphosphates. Among all the compounds, cytidine 5′-O-α,β-methylene-β-triphosphate inhibited RNase H activity of HIV-1 reverse transcriptase with a Ki value of 225 μM.
Co-reporter:Bhupender S. Chhikara, Deendayal Mandal, Keykavous Parang
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 10) pp:4601-4608
Publication Date(Web):October 2010
DOI:10.1016/j.ejmech.2010.07.024
Cytarabine is a chemotherapeutic agent predominately used for the treatment of acute myeloid leukemia and lymphoblastic leukemia. Cytarabine is a polar nucleoside, has a short plasma half-life, and its use is associated with severe side effects. Fatty acyl derivatives of cytarabine were synthesized with the expectation to improve cellular uptake and generate derivatives with a longer duration of action. Multi-step protection and deprotection reactions of hydroxyl and amino groups and conjugation with a fatty acid (i.e., myristic acid and 12-thioethyldodecanoic acid) afforded 5′-O-substituted, 2′-O-substituted, and 2′,5′-disubstituted fatty acyl derivatives of cytarabine. 2′,5′-Dimyristoyl derivative of cytarabine was found to inhibit the growth of CCRF-CEM cells by approximately 76% at concentration of 1 μM after 96 h incubation.Cytarabine is an anti-leukemia agent with a short plasma half-life. Three classes of 5′-O-substituted, 2′-O-substituted, and 2′,5′-disubstituted fatty acyl derivatives of cytarabine were synthesized and their anti-leukemia activities were evaluated.
Co-reporter:Hitesh K. Agarwal, Anil Kumar, Gustavo F. Doncel, Keykavous Parang
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 23) pp:6993-6997
Publication Date(Web):1 December 2010
DOI:10.1016/j.bmcl.2010.09.133
Chemical conjugates between sodium cellulose sulfate (CS), displaying contraceptive and HIV-entry inhibiting properties, and nucleoside reverse transcriptase inhibitors (NRTIs) (3′-azido-2′,3′-dideoxythymidine (AZT), 3′-fluoro-2′,3′-dideoxythymidine (FLT), or 2′,3′-dideoxy-3′-thiacytidine (3TC)) were designed to simultaneously provide contraceptive and anti-HIV activity. Two linkers, acetate and succinate, were used to conjugate the nucleoside analogs with CS. The conjugates containing cellulose sulfate-acetate (CSA) (e.g., AZT–CSA and FLT–CSA) were found to be more potent than CS and other conjugates (e.g., AZT–succinate–CS, and FLT–succinate–CS). The presence of both sulfate and the acetate groups on cellulose were critical for generating maximum anti-HIV activity. In addition to showing equal potency against wild-type and multidrug resistant HIV-1, the AZT–CSA conjugate displayed significant contraceptive activity in an animal model, providing the initial proof-of-concept for the design and synthesis of dual-activity compounds based on these combinations.The synthesis and anti-HIV activities of two classes of conjugates between sodium cellulose sulfate succinate and sodium cellulose sulfate acetate as HIV-entry inhibitors and nucleoside reverse transcriptase inhibitors are reported.
Co-reporter:Guofeng Ye, Anju Gupta, Robert DeLuca, Keykavous Parang, Geoffrey D. Bothun
Colloids and Surfaces B: Biointerfaces 2010 Volume 76(Issue 1) pp:76-81
Publication Date(Web):1 March 2010
DOI:10.1016/j.colsurfb.2009.10.016
The effects of a series of low molecular weight water-soluble cationic linear peptide analogs (LPAs, <1000 MW) with increasing hydrophobic/hydrophilic balance on lipid bilayer phase behavior and permeability were examined using liposomes composed of zwitterionic dipalmitoylphosphatidylcholine (DPPC) and mixed zwitterionic/anionic DPPC/dipalmitoylphosphatidylglycerol (DPPG) lipid bilayers. LPAs were synthesized using a previously reported alkyl linkage strategy as Arg-Cn-Arg-Cn-Lys, where Cn represents the saturated alkyl linkage separating the cationic residues (n = 4, 7, or 11) (Ye et al., 2007 [1]). Differential scanning calorimetry results show that the cationic LPAs bound to and disrupted DPPC and, to a greater extent, DPPC/DPPG phase behavior. When added to preformed unilamellar liposomes, the LPAs led to significant structural changes based on cryogenic transmission electron microscopy (cryo-TEM). Coupling cryo-TEM with carboxyfluorescein leakage studies indicate that the LPAs induced permeabilization through bilayer expansion, which caused membrane thinning. The effects were inconsistent with increasing LPA hydrophobicity, which suggests that a cooperative effect between electrostatic binding and hydrophobic insertion determined the location of LPAs within the bilayer and their membrane activity. Our results for LPA-induced membrane disruption correlate with previous breast cancer cell uptake studies that showed minimal LPA-C4 uptake, but high LPA-C11 uptake through a non-endocytic mechanism.
Co-reporter:Yousef Ahmadibeni, Rakesh K. Tiwari, Gongqin Sun and Keykavous Parang
Organic Letters 2009 Volume 11(Issue 10) pp:2157-2160
Publication Date(Web):April 13, 2009
DOI:10.1021/ol900320r
Chloromethyl polystyrene resin was reacted with 5-hydroxysalicylaldehyde in the presence of potassium carbonate to afford polymer-bound 2-hydroxybenzaldehyde. Subsequent reduction with borane solution produced polymer-bound 2-hydroxybenzyl alcohol. The reaction of immobilized 2-hydroxybenzyl alcohol with appropriate phosphitylating reagents yielded solid-phase cycloSaligenyl mono-, di-, and triphosphitylating reagents, which were reacted with unprotected nucleosides, followed by iodine oxidation, deprotection of cyanoethoxy groups, and the basic cleavage, respectively, to afford 5′-O-nucleoside mono-, di-, and triphosphoramidates in 52−73% overall yield.
Co-reporter:Guofeng Ye, Aaron D. Schuler, Yousef Ahmadibeni, Joel R. Morgan, Absar Faruqui, Kezhen Huang, Gongqin Sun, John A. Zebala, Keykavous Parang
Bioorganic Chemistry 2009 Volume 37(Issue 4) pp:133-142
Publication Date(Web):August 2009
DOI:10.1016/j.bioorg.2009.05.003
Phosphopeptide pTyr-Glu-Glu-Ile (pYEEI) has been introduced as an optimal Src SH2 domain ligand. Peptides, Ac-K(IDA)pYEEIEK(IDA) (1), Ac-KpYEEIEK (2), Ac-K(IDA)pYEEIEK (3), and Ac-KpYEEIEK(IDA) (4), containing 0–2 iminodiacetate (IDA) groups at the N- and C-terminal lysine residues were synthesized and evaluated as the Src SH2 domain binding ligands. Fluorescence polarization assays showed that peptide 1 had a higher binding affinity (Kd = 0.6 μM) to the Src SH2 domain when compared with Ac-pYEEI (Kd = 1.7 μM), an optimal Src SH2 domain ligand, and peptides 2–4 (Kd = 2.9–52.7 μM). The binding affinity of peptide 1 to the SH2 domain was reduced by more than 2-fold (Kd = 1.6 μM) upon addition of Ni2+ (300 μM), possibly due to modest structural effect of Ni2+ on the protein as shown by circular dichroism experimental results. The binding affinity of 1 was restored in the presence of EDTA (300 μM) (Kd = 0.79 μM). These studies suggest that peptides containing IDA groups may be used for designing novel SH2 domain binding ligands.Phosphopeptides containing 0–2 iminodiacetate groups at the N- and C-terminal lysine residues were designed for studying the Src SH2 domain interactions.
Co-reporter:Rakesh Tiwari Dr. Dr.
ChemBioChem 2009 Volume 10( Issue 15) pp:2445-2448
Publication Date(Web):
DOI:10.1002/cbic.200900462
Co-reporter:Suman Penugonda;Anil Kumar;Hitesh K. Agarwal;Reza Mehvar
Journal of Pharmaceutical Sciences 2008 Volume 97( Issue 7) pp:2649-2664
Publication Date(Web):
DOI:10.1002/jps.21161
Abstract
To control the rate of release of methylprednisolone (MP) in lysosomes, new dextran–MP conjugates with peptide linkers were synthesized and characterized. Methylprednisolone succinate (MPS) was attached to dextran 25 kDa using linkers with 1–5 Gly residues. The release characteristics of the conjugates in pH 4.0 and 7.4 buffers, blood, liver lysosomes, and various lysosomal proteinases were determined using a size-exclusion and/or a newly developed reversed-phase HPLC method capable of simultaneous quantitation of MP, MPS, and all five possible MPS-peptidyl intermediates. We synthesized conjugates with ≥90% purity and 6.9–9.5% (w/w) degree of MP substitution. The conjugates were stable at pH 4.0, but released MP and intact MPS-peptidyl intermediates in the pH 7.4 buffer and rat blood, with faster degradation rates for longer linkers. Rat lysosomal fractions degraded the conjugates to MP and all the possible intermediates also at a rate directly proportional to the length of the peptide. Whereas the degradation of the conjugates by cysteine peptidases (papain or cathepsin B) was relatively substantial, no degradation was observed in the presence of aspartic (cathepsin D) or serine (trypsin) proteinases, which do not cleave peptide bonds with Gly. These newly developed dextran conjugates of MP show promise for controlled delivery of MP in lysosomes. © 2007 Wiley-Liss, Inc. and the American Pharmacists Association J Pharm Sci 97:2649–2664, 2008
Co-reporter:Krishna C. Chimalakonda, Hitesh K. Agarwal, Anil Kumar, Keykavous Parang and Reza Mehvar
Bioconjugate Chemistry 2007 Volume 18(Issue 6) pp:2097
Publication Date(Web):October 9, 2007
DOI:10.1021/bc700193d
A liver-selective prodrug (3TCSD) of the antiviral drug lamivudine (3TC) was developed and characterized. 3TC was coupled to dextran (∼25 kDa) using a succinate linker, and the in vitro and in vivo behavior of the conjugate was studied using newly developed size-exclusion and reversed-phase analytical methods. Synthesized 3TCSD had a purity of >99% with a degree of substitution of 6.5 mg of 3TC per 100 mg of the conjugate. Furthermore, the developed assays were precise and accurate in the concentration ranges of 0.125–20, 0.36–18, and 1–50 µg/mL for 3TC, 3TC succinate (3TCS), and 3TCSD, respectively. In vitro, the conjugate slowly released 3TC in the presence of rat liver lysosomes, whereas it was stable in the corresponding buffer. In vivo in rats, conjugation of 3TC to dextran resulted in 40- and 7-fold decreases in the clearance and volume of distribution of the drug, respectively. However, the accumulation of the conjugated 3TC in the liver was 50-fold higher than that of the parent drug. The high accumulation of the conjugate in the liver was associated with a gradual and sustained release of 3TC in the liver. These studies indicate the feasibility of the synthesis of 3TCS–dextran and its potential use for the selective delivery of 3TC to the liver.
Co-reporter:Yousef Ahmadibeni Dr.
Angewandte Chemie 2007 Volume 119(Issue 25) pp:
Publication Date(Web):11 MAY 2007
DOI:10.1002/ange.200605029
Brückenbauer: Oligodesoxynucleotide (ODNs) mit Diphosphodiester-Brücken wurden durch Festphasensynthese hergestellt. Die modifizierten ODNs bilden mit komplementären modifizierten wie nichtmodifizierten Nucleinsäuren stabile Duplexe. Die modifizierten Oligomere wurden durch DNase I und 3′-Exonuclease I unter Bedingungen, bei denen nichtmodifizierte ODNs abgebaut werden, nicht abgebaut. B=T, A, G oder C.
Co-reporter:Anil Kumar Dr.;Yuehao Wang Dr.;Xiaofeng Lin Dr.;Gongqin Sun
ChemMedChem 2007 Volume 2(Issue 9) pp:
Publication Date(Web):25 MAY 2007
DOI:10.1002/cmdc.200700074
3-Phenylpyrazolo[3,4-d]pyrimidine (PhPP) derivatives substituted with an alkyl or aryl carboxylic acid at the N1-endocyclic amine, such as PhPP-CH2COOH (IC50=250 μM), and peptides Ac-CIYKYY (IC50=400 μM) and Ac-YIYGSFK (IC50=570 μM) were weak inhibitors of polyE4Y phosphorylation by active c-Src. A series of PhPP–peptide conjugates were synthesized using PhPP as an ATP mimic and CIYKYY or YIYGSFK as a peptide substrate to improve the inhibitory potency against active c-Src kinase. PhPP derivatives were attached to the N terminus or the side chain of amino acids in the peptide template. Two N-terminal substituted conjugates, PhPP-CH2CO-CIYKYY (IC50=0.38 μM) and PhPP-CH2CO-YIYGSFK (IC50=2.7 μM), inhibited the polyE4Y phosphorylation by active c-Src significantly higher than that of the parent compounds. The conjugation of PhPP with the peptides produced a synergistic inhibition effect possibly through creation of favorable interactions between the conjugate and the kinase domain as shown by molecular modeling studies.
Co-reporter:Yousef Ahmadibeni Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 25) pp:
Publication Date(Web):11 MAY 2007
DOI:10.1002/anie.200605029
Build a bridge: Oligodeoxynucleotides (ODNs) containing diphosphodiester bridges were synthesized by a solid-phase synthesis strategy. Modified ODNs formed stable duplexes with complementary modified and unmodified nucleic acid sequences. The modified oligomers were resistant to degradation by DNase I and 3′-exonuclease I under conditions in which unmodified ODNs were degraded. B=T, A, G, or C.
Co-reporter:Guofeng Ye, Marina Ayrapetov, Nguyen-Hai Nam, Gongqin Sun, Keykavous Parang
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 22) pp:4994-4997
Publication Date(Web):15 November 2005
DOI:10.1016/j.bmcl.2005.08.005
Two solid-phase binding assays were designed and evaluated for their potential use in comparing the affinity of peptides to the Src SH2 domain. Resin beads attached to peptides were incubated with the enhanced green fluorescence protein(EGFP)-Src SH2 domain fusion protein or the biotinylated Src SH2 domain and extensively washed. The beads-attached tetrapeptides with high affinities to the EGFP-Src SH2 domain showed more fluorescence intensity than those beads containing tetrapeptides with weak binding affinities, as shown by fluorescence microscopy and fluorescence imaging system. Only the beads attached to pYEEI produced a dark purple color on incubation of the beads, respectively, with the biotinylated Src kinases SH2 domain, alkaline phosphatase-coupled streptavidin, and BCIP/NBT. These solid-phase binding assays may have potential applications for the screening of peptides for the Src kinases SH2 domains.Two solid-phase binding assays were evaluated for potential use in the screening of peptides as the Src SH2 domain inhibitors.
Co-reporter:Nguyen-Hai Nam, Rebecca L Pitts, Gongqin Sun, Soroush Sardari, Amie Tiemo, Mingxing Xie, Bingfang Yan, Keykavous Parang
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 4) pp:779-787
Publication Date(Web):15 February 2004
DOI:10.1016/j.bmc.2003.10.060
Src homology-2 (SH2) domains are noncatalytic motifs containing approximately 100 amino acid residues that are involved in intracellular signal transduction. The phosphotyrosine-containing tetrapeptide pTyr-Glu-Glu-Ile (pYEEI) binds to Src SH2 domain with high affinity (Kd=100 nM). The development of five classes of tetrapeptides as inhibitors for the Src SH2 domain is described. Peptides were prepared via solid-phase peptide synthesis and tested for affinity to Src SH2 domain using a fluorescence polarization based assay. All of the N-terminal substituted pYEEI derivatives (class II) presented binding affinity (IC50=of 2.7–8.6 μM) comparable to pYEEI (IC50=6.5 μM) in this assay. C-Terminal substituted pYEEI derivatives (class III) showed a lower binding affinity with IC50 values of 34–41 μM. Amino-substituted phenylalanine derivatives (class IV) showed weak binding affinities (IC50=16–153 μM). Other substitutions on phenyl ring (class I) or the replacement of the phenyl ring with other cyclic groups (class V) dramatically decreased the binding of tetrapeptides to Src SH2 (IC50>100 μM). The ability of pYEEI and several of the tetrapeptides to inhibit the growth of cancer cells were assessed in a cell-based proliferation assay in human embryonic kidney (HEK) 293 tumor cells. The binding affinity of several of tested compounds against Src SH2 domain correlates with antiproliferative activity in 293T cells. None of the compounds showed any significant antifungal activity against Candida albicans ATCC 14053 at the maximum tested concentration of 10 μM. Overall, these results provided the structure–activity relationships for some FEEI and YEEI derivatives designed as Src SH2 domain inhibitors.Five distinct classes of tetrapeptides were synthesized and tested for affinity to Src SH2 domain. Design of tetrapeptides focused on several features: substitutions on the tyrosine or phenylalanine phenyl ring; N- and C-terminal substitution; phosphate group replacement; and tyrosine replacement with other cyclic groups.
Co-reporter:Nguyen-Hai Nam, Sungsoo Lee, Guofeng Ye, Gongqin Sun, Keykavous Parang
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 22) pp:5753-5766
Publication Date(Web):15 November 2004
DOI:10.1016/j.bmc.2004.08.043
A number of Src SH2 domain inhibitors enhance the kinase catalytic activity by switching the closed inactive to the open active conformation. ATP-phosphopeptide conjugates were designed and synthesized as Src tyrosine kinase inhibitors based on a tetrapeptide sequence pTyr-Glu-Glu-Ile (pYEEI) and ATP to block the SH2 domain signaling and substrate phosphorylation by ATP, respectively. In general, ATP-phosphopeptide conjugates with optimal linkers such as compounds 5 and 7 (Ki = 1.7–2.6 μM) showed higher binding affinities to the ATP-binding site relative to the other ATP-phosphopeptide conjugates having short or long linkers, 1–4 and 6, (Ki = 10.1–16.1 μM) and ATP (Km = 74 μM). These ATP-phosphopeptide conjugates may serve as novel templates for designing protein tyrosine kinase inhibitors to block SH2 mediated protein–protein interactions and to counter the activation of enzyme that resulted from the SH2 inhibition.ATP-phosphopeptide conjugates were synthesized and evaluated in vitro against c-Src and Lck.
Co-reporter:Y. Ahmadibeni, C. Dash, M. J. Hanley, S. F. J. Le Grice, H. K. Agarwal and K. Parang
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 6) pp:NaN1274-1274
Publication Date(Web):2010/01/13
DOI:10.1039/B922846B
A polymer-bound α,β-methylene-β-triphosphitylating reagent was synthesized and subjected to reactions with unprotected nucleosides, followed by oxidation, deprotection of cyanoethoxy groups, and acidic cleavage to afford nucleoside 5′-O-α,β-methylene-β-triphosphates. Among all the compounds, cytidine 5′-O-α,β-methylene-β-triphosphate inhibited RNase H activity of HIV-1 reverse transcriptase with a Ki value of 225 μM.

![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)