•The order of stability of functionalized Nbn + 1Cn MXenes is Nbn + 1CnO2 > Nbn + 1Cn(OH)2 > Nbn + 1CnF2 > Nbn + 1Cn(OCH3)2.•Bare Nb2C MXene sheets were theoretically predicted to have high quantum capacitances of 1860 and 1111 F/g at negative and positive electrodes, respectively.•F-, O-, OH- and OCH3 functionalization decreases the quantum capacitance of the Nbn + 1Cn MXenes at anode.•OCH3-functionalized Nb2C MXene sheets are suitable for the field emitters with an extremely low work function near 1.0 eV.To explore applicability of MXenes in supercapacitors and electronic devices, the quantum capacitances and work functions of Nbn + 1CnZ2 (n = 1, 2, 3 and 4, and Z = bare, F, O, OH, OCH3), were investigated using ab initio density functional theory. We demonstrate that both bare and functionalized niobium carbides break through the quantum capacitance limitation of graphene-based electrodes of supercapacitors. Although the functional groups substantially decrease the quantum capacitances of bare MXene sheets at positive electrodes, they have a relatively small influence at negative electrodes except for the Nb2C sheet. The theoretical quantum capacitance of bare MXene sheets at the positive electrode decreases with the increase of the layer thickness. The introduction of F and O atoms makes the work function increase and both OH and OCH3 groups lower the work function of the MXene sheets. Our work demonstrates that bare Nb2C MXene sheet is a promising material for electrodes of supercapacitors with theoretical quantum capacitances of 1828.4 and 1091.1 F/g at positive and negative electrodes, respectively, and OCH3-functionalized Nbn + 1Cn MXene sheets are suitable for the field emitters with an ultralow work function of 1.0 eV.Download high-res image (153KB)Download full-size image
The data reported in this article are structural and physicochemical properties for bare and F, O, OH and CH3O-functionalized Nbn+1Cn (n = 1, 2, 3 and 4) MXenes. The structural properties are presented as top views and side views from the X direction of the optimal structures of studied MXenes. The physicochemical properties include quantum capacitances, electrostatic potentials and electronic properties such as the projected density of states (PDOS) and band structures. Further interpretation and discussion of these data can be obtained from the article entitled “Possibility of bare and functionalized niobium carbide MXenes for electrode materials of supercapacitors and field emitters” (Xin and Yu, 2017) [1].
The Journal of Physical Chemistry C 2016 Volume 120(Issue 10) pp:5288-5296
Publication Date(Web):March 4, 2016
DOI:10.1021/acs.jpcc.5b10366
Sodium storage capacity, mobility, and volume change during sodiation on the surfaces of interlayer-expanded Ti3C2 MXenes are investigated using ab initio density functional theory. The theoretical results reveal that the interlayer-expanded bare, F-, O-, and OH-functionalized Ti3C2 MXenes exhibit low barriers for sodium diffusion and small changes of lattice constant during sodiation. In addition, enlarged interlayer distance enables the stable multilayer adsorption on the bare and O-functionalized Ti3C2 MXenes and therefore significantly enhances their theoretical capacities. Both bare and O-functionalized Ti3C2 MXenes are predicted to be prospective anode materials for sodium-ion batteries with high theoretical capacities, fast discharge/charge rates, and good cycling performances. The present results provide a new route to improve the battery performances of anode materials based on MXene intercalation hosts.
Journal of Chemical & Engineering Data 2016 Volume 61(Issue 7) pp:2236-2243
Publication Date(Web):June 3, 2016
DOI:10.1021/acs.jced.5b00926
Solubility of inorganic salts in the mixed systems containing surfactants and sodium tripolyphosphate (STPP) is very important for the production of laundry detergents. In this work we explored the solubility of NaCl, Na2SO4, and Na2CO3 in aqueous surfactant and STPP solutions using an ion selective electrode at 298.15 K. In the experiments, electromotive forces (EMF) for the ternary systems were measured using an ion-meter with a sodium ion-selective composite electrode and the relationship between the solubility and EMF was analyzed by a potentiometric analysis. The proposed method has advantages in avoiding the solid–liquid separation operation and solution supersaturation. The experimental results indicated that the salting-out effect caused by surfactant and STPP can decrease the solubility of sodium salts in the aqueous systems.
Journal of Materials Chemistry A 2014 vol. 2(Issue 23) pp:8910-8917
Publication Date(Web):12 Mar 2014
DOI:10.1039/C4TA00103F
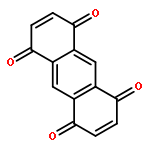
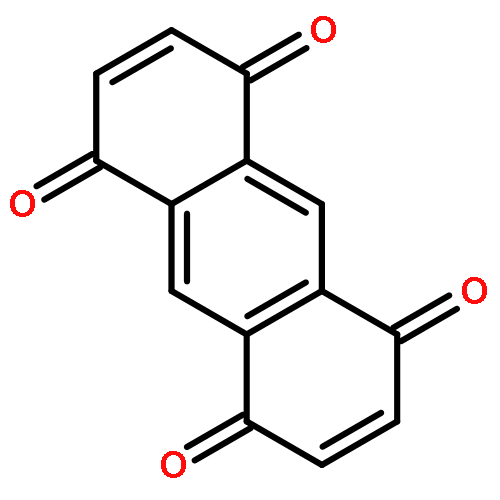
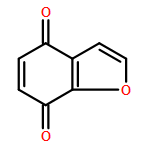
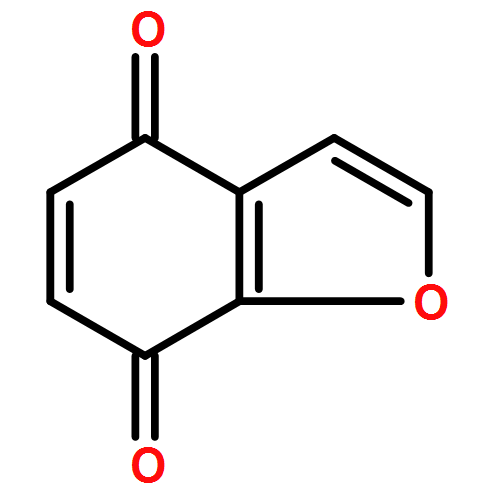
9,10-Anthraquinone (AQ) and its derivatives, i.e., benzofuro[5,6-b]furan-4,8-dione (BFFD), benzo[1,2-b:4,5-b′]dithiophene-4,8-dione (BDTD) and pyrido[3,4-g]isoquinoline-5,10-dione (PID), are environmentally friendly and cheap electrode materials. However, their significant solubility in electrolyte solutions limits the cycle performance of lithium-ion batteries. In this work a comparative investigation of these four organic molecules adsorbed on monolayer graphene and hexagonal boron nitride (h-BN) has been carried out using van der Waals (vdW) dispersion-corrected density-functional theory (DFT). The calculated results indicate that the vdW dispersion contributes to more than 80% of the total attractive interaction for all the complexes studied. The binding energies range from 1.06 to 1.31 eV, showing strong physisorption. The calculated binding energies of the four organic molecules are in the order: BFFD < BDTD < AQ < PID on monolayer graphene and BFFD < BDTD < PID < AQ on monolayer h-BN. The physisorption causes a work function shift relative to the isolated graphene or h-BN nanosheet in the order: AQ < BDTD < BFFD < PID on both the graphene and h-BN nanosheets. This sequence is dominated by the work functions of the four organic molecules. The strong physisorption suggests that the solubility of the four organic compounds in the electrolyte solutions can be reduced by binding them to a graphene or h-BN nanosheet, making the organic compound–graphene or organic compound–h-BN composite a promising electrode material for lithium-ion batteries.
Electroactive organic compounds are a novel group of green cathode materials for rechargeable metal-ion batteries. However, the organic battery life is short because the organic compounds can be dissolved by nonaqueous electrolytes. Here a comparative investigation of phenanthraquinone (PQ), pyromellitic dianhydride (PMDA) and their derivatives, i.e., benzo[1,2-b:4,3-b′]difuran-4,5-dione (BDFD), benzo[1,2-b:4,3-b′]dithiophene-4,5-quinone (BDTQ), 3,8-phenanthroline-5,6-dione (PAD), pyromellitic dithioanhydride (PMDT), pyromellitic diimide (PMDI) and 1,4,5,8-anthracenetetrone (ATO), adsorbed on graphene is performed using a density functional theory (DFT) with a van der Waals (vdW) dispersion-correction. The computed results show a strong physisorption with the binding energies between 1.10 and 1.56 eV. A sequence of the calculated binding energies from weak to strong is found to be BDFD < BDTQ < PMDA ≤ PMDI < PMDT < PQ < PAD < ATO. The formation of stable organic molecule–graphene nanocomposites can prevent the dissolution of the eight organic compounds in nonaqueous electrolyte and hence improve cycling performance of batteries. In addition, the work functions for the nanocomposites are found to be strongly affected by the work function of each organic compound. To understand the DFT results, a novel simple expression is proposed to predict the work function of the nanocomposites from the interfacial dipole and the work functions of the isolated graphene nanosheet and organic molecules. The predicted work functions for the nanocomposites from the new equation agree quite well with the values calculated from the vdW dispersion-corrected DFT.Keywords: adsorption; electroactive organic compound; first-principles; graphene; work function
Co-reporter:Yingfeng Li, Meicheng Li, Tai Wang, Fan Bai and Yang-Xin Yu
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 11) pp:5213-5220
Publication Date(Web):17 Jan 2014
DOI:10.1039/C3CP54275K
The nucleation path of graphene growth on the Cu(111) surface is investigated by importing carbon atoms step-by-step using density functional theory (DFT) calculations. An overall path of graphene nucleation has been proposed based on configuration and energy analysis. At the very first stage, linear chains will be formed and dominate the copper surface. Then, Y-type (furcate) carbon species will be shaped when new carbon atoms are absorbed aside the linear chains. Finally, ring-containing carbon species and graphene islands will be formed stepwise, with energetic preference. We find that the Y-type and ring-containing carbon species are not likely formed directly at the initial stage of graphene nucleation, but should be formed starting from linear chains. The nucleation limiting step is the formation of the Y-type species, which must pass an energy barrier of about 0.25 eV. These underlying observations are instructive to stimulate future experimental efforts on graphene synthesis.
The Journal of Physical Chemistry C 2014 Volume 118(Issue 48) pp:28274-28282
Publication Date(Web):November 20, 2014
DOI:10.1021/jp5095195
Low contact barrier electrodes and various field-emitting devices require a tunable work function, and graphene is a dream material for these applications. In this work, the theoretical investigations on the variation of the work function for monolayer graphene doped with different kinds of atoms from groups IIA–VIA of the Periodic Table are reported. The geometry, density of states, dipole moment, and work function of each heteroatom-doped graphene are calculated using ab initio density functional theory with a dispersion correction. The obtained formation energy of the heteroatom-doped graphenes is in the order: N < B < P < O < S < Si < As < Se < Ge < Al < Ga. The work functions without an electric field abide by a periodic law in terms of doping atoms except for O-doped graphene. The calculated results demonstrate that the work functions of all heteroatom-doped graphenes are a linear function of the applied external electric field intensity, and the slopes of the lines deviate from the ideal value to a different extent, which is mainly dependent on the polarization of the heteroatom–carbon bonds and the production of the induced dipole moments of the doped graphenes. The present calculated results make it known that the graphene work function can be tinkered up from 0.5 to 8.5 eV by using different kinds of doping atoms of group IIIA–VIA elements and applying an electric field with various intensities. Such a wide range of adjustable work function makes graphene a very promising material for contact electrodes and field-emitting devices.
Journal of Materials Chemistry A 2013 vol. 1(Issue 43) pp:13559-13566
Publication Date(Web):30 Aug 2013
DOI:10.1039/C3TA12639K
It is known that low-dimensional carbon allotropes can be used as a new class of anode materials for lithium-ion batteries. However, the existing carbon allotropes cannot meet the increasing energy and power demand, and thus there is still a need for further development of new materials for lithium-ion batteries. In the present work, a new graphene allotrope, known as graphenylene, is found to be capable of storing lithium with greater density of energy. Ab initio density functional theory calculations indicate that the unique dodecagonal holes in graphenylene enable lithium ions to diffuse both on and through graphenylene layers with energy barriers no higher than 0.99 eV. Adsorption of a lithium atom on graphenylene is stronger than that on pristine graphene. The highest lithium storage capacities for monolayer and bilayer graphenylene compounds are Li3C6 and Li2.5C6, respectively, which correspond to specific capacities of 1116 and 930 mA h g−1. Both specific and volumetric capacities of lithium-intercalated graphenylene compounds are significantly larger than those for graphene. The high lithium mobility and large lithium storage capacity demonstrate that graphenylene is a promising anode material for modern lithium-ion batteries.
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 39) pp:16819-16827
Publication Date(Web):13 Aug 2013
DOI:10.1039/C3CP51689J
The electronic and adsorption properties of graphene can be changed significantly through substitutional doping with nitrogen and nitrogen decoration of vacancies. Here ab initio density functional theory with a dispersion correction was used to investigate the stability, magnetic and adsorption properties of nine defects in graphene, including both nitrogen substitutional doping and nitrogen decoration of vacancies. The results indicate that only pyridinic N2V2 defect in graphene shows a ferromagnetic spin structure with high magnetic moment and magnetic stabilization energy. Not all nitrogen-doped defects can improve the capacity of the lithium-ion batteries. The adsorption energies of a lithium atom on nitrogen-substituted graphenes are more positive, indicating that they are meta-stable and no better than the pristine graphene as anode materials of lithium-ion batteries. Nitrogen-decorated single and double vacancy defects, especially for the pyridinic N2V2 defect in graphene, can greatly improve the reversible capacity of the battery in comparison with the pristine graphene. The theoretical prediction of the reversible capacity of the battery is 1039 mA h g−1 for the nitrogen-doped graphene material synthesized by Wu et al., which is in good agreement with the experimental data (1043 mA h g−1). The theoretical computations suggest that nitrogen-decorated single and double vacancy defects in graphene are the promising candidate for anode materials of lithium-ion batteries. Each nitrogen atom in the decoration can improve the reversible capacity of the battery by 63.3–124.5 mA h g−1 in a 4 × 4 supercell of graphene. The present work provides crucial information for the development of N-doped graphene-based anode materials of lithium-ion batteries.
Science China Chemistry 2013 Volume 56( Issue 4) pp:524-532
Publication Date(Web):2013 April
DOI:10.1007/s11426-012-4825-1
Unrestrained molecular dynamics (MD) simulations have been carried out to characterize the stability of DNA conformations and the dynamics of A-DNA→B-DNA conformational transitions in aqueous RbCl solutions. The PARM99 force field in the AMBER8 package was used to investigate the effect of RbCl concentration on the dynamics of the A→B conformational transition in the DNA duplex d(CGCGAATTCGCG)2. Canonical A- and B-form DNA were assumed for the initial conformation and the final conformation had a length per complete turn that matched the canonical B-DNA. The DNA structure was monitored for 3.0 ns and the distances between the C5′ atoms were obtained from the simulations. It was found that all of the double stranded DNA strands of A-DNA converged to the structure of B-form DNA within 1.0 ns during the unrestrained MD simulations. In addition, increasing the RbCl concentration in aqueous solution hindered the A→B conformational transition and the transition in aqueous RbCl solution was faster than that in aqueous NaCl solution for the same electrolyte strength. The effects of the types and concentrations of counterions on the dynamics of the A→B conformational transition can be understood in terms of the variation in water activity and the number of accumulated counterions in the major grooves of A-DNA. The rubidium ion distributions around both fixed A-DNA and B-DNA were obtained using the restrained MD simulations to help explain the effect of RbCl concentration on the dynamics of the A→B conformational transition.
Science China Chemistry 2013 Volume 56( Issue 12) pp:1735-1742
Publication Date(Web):2013 December
DOI:10.1007/s11426-013-4959-9
DNA and its conformational transition can be used to design nanometer-scale structures, nano-tweezers and nanomechanical devices. Experiments and molecular simulations have been used to study the concentration effect on the A-DNA→B-DNA conformational transition, but a systematical investigation on counterion effect on the dynamics of this transition has not been reported up to now. In present work, restrained and unrestrained molecular dynamics (MD) simulations have been performed to characterize the stability of DNA conformations and the dynamics of A-DNA→B-DNA transitions in aqueous solutions with different alkali metal counterions. The DNA duplex d(CGCGAATTCGCG)2, coion Cl− and counterions Li+, Na+, K+, Rb+ and Cs+ as well as water molecule were considered using the PARM99 force field in the AMBER8 package. It was found that B-form DNA is more stable than A-form DNA in aqueous electrolyte solutions with different alkali metal counterions. Increasing KCl concentration in solution hinders the A-DNA→B-DNA transition and the transition times for different alkali metal counterions conform to neither the simple sequence related to naked ion size nor to hydrated diameter, but an apparently abnormal sequence of K+ < Rb+ < Cs+ < Na+ < Li+. This abnormal sequence can be well understood in terms of an electrostatic model based on the effective cation diameters and the modified mean-spherical approximation (MMSA). The present results provide valuable information for the design of DNA-based nanomaterials and nanodevices.
Adsorption of carbon monoxide on different Ag+-exchange sites of Ag–ZSM-5 zeolite has been investigated using density functional theory. The coordination and local geometry of the Ag+ ion in Ag–ZSM-5 as well as adsorption structures and energies of CO adsorbed on these sites are explored extensively. The structure of Ag+-exchange sites, location of the Al atom on the T site, and number of the Al atoms contained in the sites are considered in the theoretical calculations. The calculated results show that the AgO coordination number of two is strongly preferred before and after CO adsorption. The AgO bond lengths are in a broad range of 2.2–2.9 Å, and the AgC bond lengths for CO adsorbed on Ag–ZSM-5 zeolite are calculated to be 2.0–2.2 Å. Both AgO and AgC bond lengths for CO–Ag–ZSM-5 complex are longer than those for CO–Cu–ZSM-5 complex. The calculated adsorption energy of CO adsorbed on the I2 sites is between 28.5 and 29.6 kcal/mol, and that on the Z5, Z6, M5 and M6 sites containing one Al atom on the T position is between 11.3 and 18.9 kcal/mol whereas the calculated adsorption energy of CO adsorbed on the M7 site containing one Al atom is 19.9 kcal/mol. The introduction of the two Al atoms to the Ag+-exchange site results in a reduction of CO adsorption energy. In general, the adsorption energy of CO on Ag–ZSM-5 is lower than that on Cu–ZSM-5. The predicted coordination of the Ag+ ion, bond lengths of AgO and AgC as well as adsorption energy are in accord with available experimental results.
Co-reporter:Yuan-Xiang Zheng, Yang-Xin Yu, and Ying-Feng Li
Industrial & Engineering Chemistry Research 2011 Volume 50(Issue 10) pp:6460
Publication Date(Web):April 6, 2011
DOI:10.1021/ie102379h
Based on the thermodynamic perturbation theory for polymer, a new equation is proposed by incorporating a wide range of molecular flexibility and perturbative interactions to describe the isotropic–anisotropic (nematic) phase transition phenomena of the semiflexible polymers. In the new equation, the framework of the Helmholtz free energy of the system is the same as the Onsager-like theory. The entropy loss due to the orientation is estimated by the Khokhlov–Semenov (KS) theory. With regard to the configurational free energy, the polymer is envisioned as a series of subchains. The Parsons–Lee approximation is used to account for the higher virial coefficients of the subchain and Yu equation for the hard-sphere-chain fluid is adopted to modify the defect of the first order approximation in dealing with the associating points. An analytical expression of the perturbative term is obtained by employing the “square peg in a round hole” potential function and the mean-field approximation. The hard-core part of the equation reduces to the Dupre–Yang theory when the stiffness of the molecule is high. When the model approaches the limit of the random coil, a modified equation of the hard-sphere-chain fluid is obtained. The present theory has been used to predict the isotropic–nematic phase equilibrium for real semiflexible polymers with two adjustable parameters. The agreements between the theoretical results and experimental data are much better than that of previous theories.
In this work, we relate the self-diffusion coefficient to the residual entropy of the system according to the free volume theory and scaling principle. The viscosity equation for a freely jointed Lennard-Jones chain fluid is then obtained from the expression of self-diffusion coefficient by applying the Stokes–Einstein equation. The real polyatomic compounds are modeled as chains of tangent Lennard-Jones segments. The segment size and energy parameter as well as chain length (expressed by the number of segments) are obtained from the experimental viscosity data. The proposed viscosity equation reproduces the experimental viscosity data with an average absolute deviation of 5.12% for 18 polyatomic compounds (1600 data points) over wide ranges of temperature and pressure. For engineering applications, the generalized model parameters for normal alkanes with the number of carbon atoms n > 3 are proposed. The segment energy parameter is suggested to be evaluated from the critical temperature, and the segment size parameter and chain length are correlated with the number of carbon atoms in an alkane molecule.
Physical Chemistry Chemical Physics 2009 vol. 11(Issue 41) pp:9382-9390
Publication Date(Web):19 Aug 2009
DOI:10.1039/B911901A
A very simple and accurate approach is proposed to predict the high-order virial coefficients of hard spheres and hard disks. In the approach, the nth virial coefficient Bn is expressed as the sum of nD−1 and a remainder, where D is the spatial dimension of the system. When n≥ 3, the remainders of the virials can be accurately expressed with Padé-type functions of n. The maximum deviations of predicted B5–B10 for the two systems are only 0.0209%–0.0044% and 0.0390%–0.0525%, respectively, which are much better than the numerous existing approaches. The virial equation based on the predicted virials diverges when packing fraction η = 1. With the predicted virials, the compressibility factors of hard sphere system can be predicted very accurately in the whole stable fluid region, and those in the metastable fluid region can also be well predicted up to η = 0.545. The compressibility factors of hard disk fluid can be predicted very accurately up to η = 0.63. The simulated B7 and B10 for hard spheres are found to be inconsistent with the other known virials and therefore they are modified as 53.2467 and 105.042, respectively.
The Journal of Physical Chemistry B 2008 Volume 112(Issue 48) pp:15407-15416
Publication Date(Web):November 11, 2008
DOI:10.1021/jp805697p
A new density functional theory (DFT) for an inhomogeneous 12−6 Lennard-Jones fluid is proposed based on the modified fundamental measure theory for repulsive interaction and a weighted density functional for attractive interaction. The Helmholtz free energy functional for the attractive part is constructed using the modified Benedict−Webb−Rubin equation of state with a mean-field weight function. Comparisons of the theoretical results with molecular simulation data suggest that the new DFT yields accurate bulk surface tension, density distributions, adsorption−desorption isotherms, pore pressures, and capillary phase transitions for the Lennard-Jones fluid confined in slitlike pores with different widths and solid-fluid interactions. The new DFT reproduces well the vapor−liquid critical temperatures of the confined Lennard-Jones fluid, whereas the mean-field theory always overestimates the critical temperatures. Because the new DFT is computationally as simple and efficient as the mean-field theory, it will provide a good reference for further development of a statistical-thermodynamic theory of complex fluid under both homogeneous and inhomogeneous conditions when disperse force has to be considered.
A density functional theory (DFT) constructed from the modified fundamental-measure theory and the modified Benedict−Webb−Rubin equation of state is presented. The Helmholtz free energy functional due to attractive interaction is expressed as a functional of attractive weighted-density in which the weight function is a mean-field-like type. An obvious advantage of the present theory is that it reproduces accurate bulk properties such as chemical potential, bulk pressure, vapor−liquid interfacial tension, and so forth when compared with molecular simulations and experiments with the same set of molecular parameters. Capabilities of the present DFT are demonstrated by its applicability to adsorption of argon and nitrogen on, respectively, a model cylindrical pore and mesoporous MCM-41 materials. Comparison of the theoretical results of argon in the model cylindrical pore with those from the newly published molecular simulations indicates that the present DFT predicts accurate average densities in the pore, slightly overestimates the pore pressure, and correctly describes the effect of the fluid−pore wall interaction on average densities and pressures in the pore. Application to adsorption of nitrogen on MCM-41 at 77.4 K shows that the present DFT predicts density profiles and adsorption isotherms in good agreement with those from molecular simulations and experiments. In contrast, the hysteresis loop of adsorption calculated from the mean-field theory shifts toward the low pressure region because a low bulk saturated pressure is produced from the mean-field equation of state. The present DFT offers a good way to describe the adsorption isotherms of porous materials as a function of temperature and pressure.
The ion density profiles and mean electrostatic potentials around DNA from the Monte Carlo simulations are compared with the predictions of the Poisson–Boltzmann equation and the density functional theory. The DNA molecules are modeled as a charged cylinder while the ions are represented as charged hard spheres with different diameter. In the density functional theory, the Helmholtz free energy functional due to hard-sphere repulsion and electrostatic interaction are obtained from the modified fundamental measure theory and a quadratic functional Taylor expansion, respectively. The results show that due to the inclusion of the ion–ion correlation, the density functional theory is more accurate than the Poisson–Boltzmann equation. The density functional theory gives accurate ion structures and mean electrostatic potentials near the surface of DNA. When the established density functional theory is combined with the cell model, the osmotic coefficients of aqueous DNA–electrolyte solutions can be predicted. The results show that the DFT-cell model captures the essential features of the experimental osmotic coefficient, but fails to give a quantitative description. Possible reasons for this discrepancy are discussed.
Co-reporter:Yang-Xin Yu, Ai-Wei Tian and Guang-Hua Gao
Physical Chemistry Chemical Physics 2005 vol. 7(Issue 12) pp:2423-2428
Publication Date(Web):20 May 2005
DOI:10.1039/B500371G
A new method to predict concentration dependence of collective diffusion coefficient of bovine serum albumin (BSA) in aqueous electrolyte solution is developed based on the generalized Stokes–Einstein equation which relates the diffusion coefficient to the osmotic pressure. The concentration dependence of osmotic pressure is evaluated using the solution of the mean spherical approximation for the two-Yukawa model fluid. The two empirical correlations of sedimentation coefficient are tested in this work. One is for a disordered suspension of hard spheres, and another is for an ordered suspension of hard spheres. The concentration dependence of the collective diffusion coefficient of BSA under different solution conditions, such as pH and ionic strength is predicted. From the comparison between the predicted and experimental values we found that the sedimentation coefficient for the disordered suspension of hard spheres is more suitable for the prediction of the collective diffusion coefficients of charged BSA in aqueous electrolyte solution. The theoretical predictions from the hard-core two-Yukawa model coupled with the sedimentation coefficient for a suspension of hard spheres are in good agreement with available experimental data, while the hard sphere model is unable to describe the behavior of diffusion due to its neglect of the double-layer repulsive charge–charge interaction between BSA molecules.
Journal of Materials Chemistry A 2014 - vol. 2(Issue 23) pp:NaN8917-8917
Publication Date(Web):2014/03/12
DOI:10.1039/C4TA00103F
9,10-Anthraquinone (AQ) and its derivatives, i.e., benzofuro[5,6-b]furan-4,8-dione (BFFD), benzo[1,2-b:4,5-b′]dithiophene-4,8-dione (BDTD) and pyrido[3,4-g]isoquinoline-5,10-dione (PID), are environmentally friendly and cheap electrode materials. However, their significant solubility in electrolyte solutions limits the cycle performance of lithium-ion batteries. In this work a comparative investigation of these four organic molecules adsorbed on monolayer graphene and hexagonal boron nitride (h-BN) has been carried out using van der Waals (vdW) dispersion-corrected density-functional theory (DFT). The calculated results indicate that the vdW dispersion contributes to more than 80% of the total attractive interaction for all the complexes studied. The binding energies range from 1.06 to 1.31 eV, showing strong physisorption. The calculated binding energies of the four organic molecules are in the order: BFFD < BDTD < AQ < PID on monolayer graphene and BFFD < BDTD < PID < AQ on monolayer h-BN. The physisorption causes a work function shift relative to the isolated graphene or h-BN nanosheet in the order: AQ < BDTD < BFFD < PID on both the graphene and h-BN nanosheets. This sequence is dominated by the work functions of the four organic molecules. The strong physisorption suggests that the solubility of the four organic compounds in the electrolyte solutions can be reduced by binding them to a graphene or h-BN nanosheet, making the organic compound–graphene or organic compound–h-BN composite a promising electrode material for lithium-ion batteries.
Journal of Materials Chemistry A 2013 - vol. 1(Issue 43) pp:NaN13566-13566
Publication Date(Web):2013/08/30
DOI:10.1039/C3TA12639K
It is known that low-dimensional carbon allotropes can be used as a new class of anode materials for lithium-ion batteries. However, the existing carbon allotropes cannot meet the increasing energy and power demand, and thus there is still a need for further development of new materials for lithium-ion batteries. In the present work, a new graphene allotrope, known as graphenylene, is found to be capable of storing lithium with greater density of energy. Ab initio density functional theory calculations indicate that the unique dodecagonal holes in graphenylene enable lithium ions to diffuse both on and through graphenylene layers with energy barriers no higher than 0.99 eV. Adsorption of a lithium atom on graphenylene is stronger than that on pristine graphene. The highest lithium storage capacities for monolayer and bilayer graphenylene compounds are Li3C6 and Li2.5C6, respectively, which correspond to specific capacities of 1116 and 930 mA h g−1. Both specific and volumetric capacities of lithium-intercalated graphenylene compounds are significantly larger than those for graphene. The high lithium mobility and large lithium storage capacity demonstrate that graphenylene is a promising anode material for modern lithium-ion batteries.
Physical Chemistry Chemical Physics 2009 - vol. 11(Issue 41) pp:NaN9390-9390
Publication Date(Web):2009/08/19
DOI:10.1039/B911901A
A very simple and accurate approach is proposed to predict the high-order virial coefficients of hard spheres and hard disks. In the approach, the nth virial coefficient Bn is expressed as the sum of nD−1 and a remainder, where D is the spatial dimension of the system. When n≥ 3, the remainders of the virials can be accurately expressed with Padé-type functions of n. The maximum deviations of predicted B5–B10 for the two systems are only 0.0209%–0.0044% and 0.0390%–0.0525%, respectively, which are much better than the numerous existing approaches. The virial equation based on the predicted virials diverges when packing fraction η = 1. With the predicted virials, the compressibility factors of hard sphere system can be predicted very accurately in the whole stable fluid region, and those in the metastable fluid region can also be well predicted up to η = 0.545. The compressibility factors of hard disk fluid can be predicted very accurately up to η = 0.63. The simulated B7 and B10 for hard spheres are found to be inconsistent with the other known virials and therefore they are modified as 53.2467 and 105.042, respectively.
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 39) pp:NaN16827-16827
Publication Date(Web):2013/08/13
DOI:10.1039/C3CP51689J
The electronic and adsorption properties of graphene can be changed significantly through substitutional doping with nitrogen and nitrogen decoration of vacancies. Here ab initio density functional theory with a dispersion correction was used to investigate the stability, magnetic and adsorption properties of nine defects in graphene, including both nitrogen substitutional doping and nitrogen decoration of vacancies. The results indicate that only pyridinic N2V2 defect in graphene shows a ferromagnetic spin structure with high magnetic moment and magnetic stabilization energy. Not all nitrogen-doped defects can improve the capacity of the lithium-ion batteries. The adsorption energies of a lithium atom on nitrogen-substituted graphenes are more positive, indicating that they are meta-stable and no better than the pristine graphene as anode materials of lithium-ion batteries. Nitrogen-decorated single and double vacancy defects, especially for the pyridinic N2V2 defect in graphene, can greatly improve the reversible capacity of the battery in comparison with the pristine graphene. The theoretical prediction of the reversible capacity of the battery is 1039 mA h g−1 for the nitrogen-doped graphene material synthesized by Wu et al., which is in good agreement with the experimental data (1043 mA h g−1). The theoretical computations suggest that nitrogen-decorated single and double vacancy defects in graphene are the promising candidate for anode materials of lithium-ion batteries. Each nitrogen atom in the decoration can improve the reversible capacity of the battery by 63.3–124.5 mA h g−1 in a 4 × 4 supercell of graphene. The present work provides crucial information for the development of N-doped graphene-based anode materials of lithium-ion batteries.
Co-reporter:Yingfeng Li, Meicheng Li, Tai Wang, Fan Bai and Yang-Xin Yu
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 11) pp:NaN5220-5220
Publication Date(Web):2014/01/17
DOI:10.1039/C3CP54275K
The nucleation path of graphene growth on the Cu(111) surface is investigated by importing carbon atoms step-by-step using density functional theory (DFT) calculations. An overall path of graphene nucleation has been proposed based on configuration and energy analysis. At the very first stage, linear chains will be formed and dominate the copper surface. Then, Y-type (furcate) carbon species will be shaped when new carbon atoms are absorbed aside the linear chains. Finally, ring-containing carbon species and graphene islands will be formed stepwise, with energetic preference. We find that the Y-type and ring-containing carbon species are not likely formed directly at the initial stage of graphene nucleation, but should be formed starting from linear chains. The nucleation limiting step is the formation of the Y-type species, which must pass an energy barrier of about 0.25 eV. These underlying observations are instructive to stimulate future experimental efforts on graphene synthesis.





![Pyrido[3,4-g]isoquinoline-5,10-dione](/data/chemimg/102800/117727-15-8.png)
![Pyrido[3,4-g]isoquinoline-5,10-dione](/data/chemimg/102800/117727-15-8_b.png)
![Benzo[1,2-b:4,3-b']dithiophene-4,5-dione](/data/chemimg/3116100/24243-31-0.png)
![Benzo[1,2-b:4,3-b']dithiophene-4,5-dione](/data/chemimg/3116100/24243-31-0_b.png)


![1H,3H-BENZO[1,2-C:4,5-C']DITHIOPHENE-1,3,5,7-TETRONE](http://img.cochemist.com/ccimg/21000/20999-59-1.png)
![1H,3H-BENZO[1,2-C:4,5-C']DITHIOPHENE-1,3,5,7-TETRONE](http://img.cochemist.com/ccimg/21000/20999-59-1_b.png)