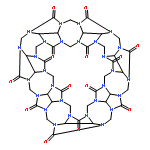
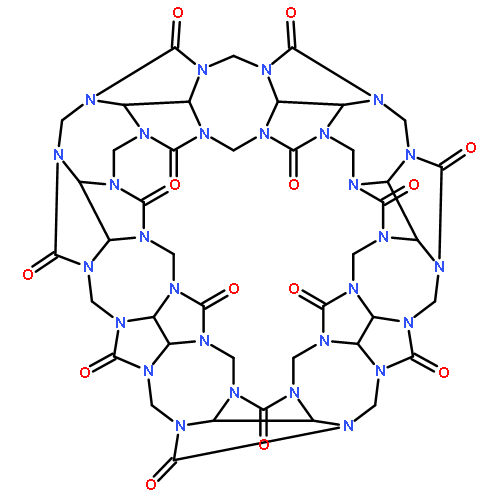
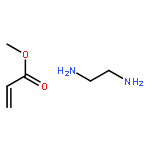
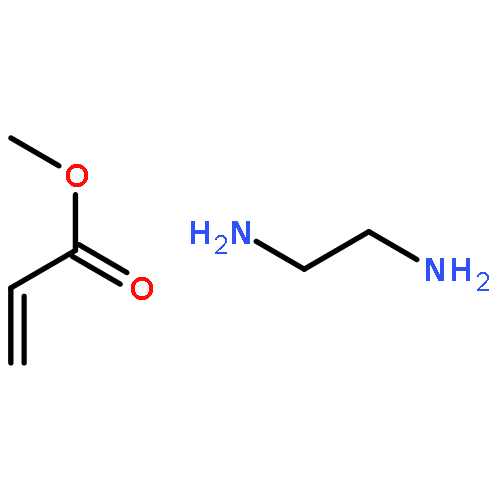
Co-reporter:Quanchen Feng, Shu Zhao, Yu Wang, Juncai Dong, Wenxing Chen, Dongsheng He, Dingsheng Wang, Jun Yang, Yuanmin Zhu, Hailiang Zhu, Lin Gu, Zhi Li, Yuxi Liu, Rong Yu, Jun Li, and Yadong Li
Journal of the American Chemical Society May 31, 2017 Volume 139(Issue 21) pp:7294-7294
Publication Date(Web):May 14, 2017
DOI:10.1021/jacs.7b01471
Improving the catalytic selectivity of Pd catalysts is of key importance for various industrial processes and remains a challenge so far. Given the unique properties of single-atom catalysts, isolating contiguous Pd atoms into a single-Pd site with another metal to form intermetallic structures is an effective way to endow Pd with high catalytic selectivity and to stabilize the single site with the intermetallic structures. Based on density functional theory modeling, we demonstrate that the (110) surface of Pm3̅m PdIn with single-atom Pd sites shows high selectivity for semihydrogenation of acetylene, whereas the (111) surface of P4/mmm Pd3In with Pd trimer sites shows low selectivity. This idea has been further validated by experimental results that intermetallic PdIn nanocrystals mainly exposing the (110) surface exhibit much higher selectivity for acetylene hydrogenation than Pd3In nanocrystals mainly exposing the (111) surface (92% vs 21% ethylene selectivity at 90 °C). This work provides insight for rational design of bimetallic metal catalysts with specific catalytic properties.
Co-reporter:Xiaofeng Yang;Aiqin Wang;Tao Zhang;Keli Han;Xiaodong Wang
The Journal of Physical Chemistry C December 10, 2009 Volume 113(Issue 49) pp:20918-20926
Publication Date(Web):2017-2-22
DOI:10.1021/jp905687g
A parallel study on silver, gold, and platinum catalysts with inert silica support is conducted both experimentally and theoretically for the liquid-phase hydrogenation of crotonaldehyde (Me−CH═CH−CH═O). We find that the silver catalyst exhibits a uniquely high selectivity toward C═O hydrogenation and the selectivity remains constant even at the conversion close to 100%. The gold catalyst, however, shows only a moderate selectivity whereas the platinum catalyst has a rather poor selectivity. Such variation in selectivity is interpreted in terms of the varied adsorption geometries of the crotonaldehyde on different metals. According to our density functional calculations of the chemisorptions of crotonaldehyde on selected M19 (M = Ag, Au, and Pt) model clusters and M(111) surface, the most favored adsorption mode for silver is the C═O oxygen atom being σ-bonded on low-coordinated silver atoms, which results in activation of the C═O bond. In contrast, so-called η4 and di-σC═C modes are preferred on Pt surface, while a πC═C adsorption mode is favored on low-coordinated gold atoms, which leads to the preference of C═C hydrogenation. Moreover, the calculations indicate that the selectivity to C═O hydrogenation is more favored on smaller silver nanoparticles. This implication has been further corroborated experimentally by investigation of silver catalysts with different particle sizes.
Co-reporter:Haohong Duan, Dongguo Li, Yan Tang, Yang He, Shufang Ji, Rongyue Wang, Haifeng Lv, Pietro P. Lopes, Arvydas P. Paulikas, Haoyi Li, Scott X. Mao, Chongmin Wang, Nenad M. Markovic, Jun Li, Vojislav R. Stamenkovic, and Yadong Li
Journal of the American Chemical Society April 19, 2017 Volume 139(Issue 15) pp:5494-5494
Publication Date(Web):March 26, 2017
DOI:10.1021/jacs.7b01376
The search for active, stable, and cost-efficient electrocatalysts for hydrogen production via water splitting could make a substantial impact on energy technologies that do not rely on fossil fuels. Here we report the synthesis of rhodium phosphide electrocatalyst with low metal loading in the form of nanocubes (NCs) dispersed in high-surface-area carbon (Rh2P/C) by a facile solvo-thermal approach. The Rh2P/C NCs exhibit remarkable performance for hydrogen evolution reaction and oxygen evolution reaction compared to Rh/C and Pt/C catalysts. The atomic structure of the Rh2P NCs was directly observed by annular dark-field scanning transmission electron microscopy, which revealed a phosphorus-rich outermost atomic layer. Combined experimental and computational studies suggest that surface phosphorus plays a crucial role in determining the robust catalyst properties.
Co-reporter:Haoxiang Xu;Cong-Qiao Xu;Daojian Cheng
Catalysis Science & Technology (2011-Present) 2017 vol. 7(Issue 24) pp:5860-5871
Publication Date(Web):2017/12/11
DOI:10.1039/C7CY00464H
Single-atom catalysts (SACs) can have high selectivity while maximizing the efficient utilization of metal atoms and are very promising for applications in catalysis. However, the design and development of SACs cannot be effectively achieved, as little theoretical effort has been directed towards exploring the activity trends for reactions on SACs. In this work, we find that there is a Brønsted–Evans–Polanyi (BEP) linear correlation between the adsorption energies of CO and O2 and transition state energies for CO oxidation on various M/MgO and M/Fs-defect MgO SACs (M = Cu, Ag, Au, Ni, Pd, and Pt) via density functional theory (DFT) calculations. Based on the contour plot of Sabatier activity from the BEP relationships and microkinetic model, we have identified the activity trends for CO oxidation on these SACs by using the adsorption energies of CO and O2 as the activity descriptors. The theoretical calculations indicate that Ag/MgO and Ag/Fs-defect MgO exhibit better catalytic performance than the other SACs. Our results provide a general picture of the identification of the activity trends for CO oxidation on MgO-supported SACs in terms of the adsorption energies of the reactants. This approach may also lay a theoretical basis for designing new SACs for reactions other than CO oxidation.
Co-reporter:Wan-Lu Li;Tian Jian;Xin Chen;Hai-Ru Li;Teng-Teng Chen;Xue-Mei Luo;Si-Dian Li;Lai-Sheng Wang
Chemical Communications 2017 vol. 53(Issue 10) pp:1587-1590
Publication Date(Web):2017/01/31
DOI:10.1039/C6CC09570D
A tubular molecular rotor B2-Ta@B18− (1) and boron drum Ta@B20− (2) with the highest coordination number of twenty in chemistry are observed via a joint photoelectron spectroscopy and first-principles theory investigation.
Co-reporter:Shu-Xian Hu;Jiwen Jian;Jing Su;Xuan Wu;Mingfei Zhou
Chemical Science (2010-Present) 2017 vol. 8(Issue 5) pp:4035-4043
Publication Date(Web):2017/05/03
DOI:10.1039/C7SC00710H
The neutral molecule NPrO and its anion NPrO− are produced via co-condensation of laser-ablated praseodymium atoms with nitric oxide in a solid neon matrix. Combined infrared spectroscopy and state-of-the-art quantum chemical calculations confirm that both species are pentavalent praseodymium nitride-oxides with linear structures that contain PrN triple bonds and PrO double bonds. Electronic structure studies show that the neutral NPrO molecule features a 4f0 electron configuration and a Pr(V) oxidation state similar to that of the isoelectronic PrO2+ ion, while its NPrO− anion possesses a 4f1 electron configuration and a Pr(IV) oxidation state. The neutral NPrO molecule is thus a rare lanthanide nitride-oxide species with a Pr(V) oxidation state, which follows the recent identification of the first Pr(V) oxidation state in the PrO2+ and PrO4 complexes (Angew. Chem. Int. Ed., 2016, 55, 6896). This finding indicates that lanthanide compounds with oxidation states of higher than +IV are richer in chemistry than previously recognized.
Co-reporter:Shu-Xian Hu;Wan-Lu Li;Liang Dong;John K. Gibson
Dalton Transactions 2017 vol. 46(Issue 36) pp:12354-12363
Publication Date(Web):2017/09/19
DOI:10.1039/C7DT02825C
For further fundamental understanding of the nature and extent of covalency in actinyl–ligand bonding, and the benefits that this may have in the design of new ligands for nuclear waste separation, there is burgeoning interest in the nature of actinyl complexes with polydentate or multiple-point-donor ligands, such as crown ethers. There are few cases of structurally authenticated molecular actinyl–crown bonds under ambient conditions. We report here the computational characterization of AnO22+–(15-crown-5) complexes, where An = U, Np, Pu, Am, and Cm, and 15-crown-5 is the cyclic polyether ligand with five ether oxygen atoms. In the gas-phase complex, the actinyl group is located inside of the crown ether, tilted slightly out of the plane of the five equatorial oxygen atoms that coordinate the actinide metal center. The actinyl–cyclic ether complexes are found to exhibit a conventional conformation, with typical An–Oaxial and An–Oequatorial distances and angles. A striking result is the enhanced stability of the insertion complex for UO22+versus NpO22+, PuO22+, AmO22+ and CmO22+, which is evaluated in the context of An–O binding strengths (esp. bonding covalency), and may have ramifications for the utility of actinyl-crown complexes in separation applications.
Co-reporter:Jian-Biao Liu;Guo P. Chen;Wei Huang;David L. Clark;W. H. Eugen Schwarz
Dalton Transactions 2017 vol. 46(Issue 8) pp:2542-2550
Publication Date(Web):2017/02/21
DOI:10.1039/C6DT03953G
Actinyl-tricarbonato anions [(AnO2)(CO3)3]4− (An = U–Cm) in various environments were investigated using theoretical approaches of quantum-mechanics, molecular-mechanics and cluster-models. Cations and solvent molecules in the 2nd coordination sphere affect the equatorial An←Oeq bonds more than the axial AnOax bonds. Common actinide contraction is found for calculated and experimental axial bond lengths of 92U to 94Pu, though no longer for 94Pu to 96Cm. The tendency of U to Pu forming actinyl(VI) species dwindles away toward Cm, which already features the preferred AnIII/LnIII oxidation state of the later actinides and all lanthanides. The well known change from d-type to typical U–Pu–Cm type and then to f-type behavior is labeled as the plutonium turn, a phenomenon that is caused by f-orbital energy-decrease and f-orbital localization with increase of both nuclear charge and oxidation state, and a non-linear variation of effective f-electron population across the actinide series. Both orbital and configuration mixing and occupation of antibonding 5f type orbitals increase, weakening the AnOax bonds and reducing the highest possible oxidation states of the later actinides.
Co-reporter:Xue-Mei Luo, Tian Jian, Long-Jiu Cheng, Wan-Lu Li, Qiang Chen, Rui Li, Hua-Jin Zhai, Si-Dian Li, Alexander I. Boldyrev, Jun Li, Lai-Sheng Wang
Chemical Physics Letters 2017 Volume 683(Volume 683) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.cplett.2016.12.051
•Bn− monoanions have been systematically investigated up to n = 30. However, B26− has remained elusive in this size range.•Here we present a joint photoelectron spectroscopy and first-principles study on the structures and bonding of this seemingly enigmatic cluster.•Extensive global minimum searches and high-level calculations reveal that isomer I dominates the experimental spectrum and represents the smallest 2D boron cluster with a hexagonal vacancy.•Isomer III is found to contribute to the measured PE spectrum as a minor species.•Chemical bonding analyses show that isomer I can be viewed as an all-boron analog of the polycyclic aromatic hydrocarbon C17H11+.Anionic boron clusters have been systematically investigated both experimentally and theoretically up to 30 atoms and have all been proved to be planar or quasi-planar (2D) in their global minima. However, the B26− cluster has remained elusive in this size range up to now, because of its complicated potential landscape. Here we present a joint photoelectron spectroscopy (PES) and first-principles study on the structures and bonding of this seemingly enigmatic cluster. Extensive global minimum searches, followed by high-level calculations and Gibbs free energy corrections, reveal that at least three 2D isomers, I (C1, 2A), II (C1, 2A), and III (C1, 2A), could contribute to the observed PE spectrum for the B26− cluster. Isomer I, which has the lowest free energy at finite temperatures, is found to dominate the experimental spectrum and represents the smallest 2D boron cluster with a hexagonal vacancy. Distinct spectral features are observed for isomer III, which has a pentagonal hole and is found to contribute to the measured PE spectrum as a minor species. Isomer II with a close-packed triangular 2D structure, which is the global minimum at 0 K, may also contribute to the observed spectrum as a minor species. Chemical bonding analyses show that the principal isomer I can be viewed as an all-boron analog of the polycyclic aromatic hydrocarbon C17H11+ in terms of the π bonds.Download high-res image (134KB)Download full-size image
Co-reporter:Xin Chen, Ya-Fan Zhao, Lai-Sheng Wang, Jun Li
Computational and Theoretical Chemistry 2017 Volume 1107(Volume 1107) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.comptc.2016.12.028
•We developed a global minimum search program named TGMin.•TGMin program is based on the basin hopping algorithm with several improvements.•TGMin program is highly effecient in finding the global minima of nanoclusters.•An overview of recent improvements and applications of TGMin are presented.Finding the global minima of nanoclusters is of great importance in cluster science and nanoscience. We have developed an efficient global minimum search program, named Tsinghua Global Minimum (TGMin, first released in 2012), based on the Basin-Hopping algorithm to find the global minima of nanoclusters, as well as periodic systems. We have recently made several improvements to the original Basin-Hopping algorithm, including a constrained perturbation function, a covalent-radius-based relaxation algorithm, an improved ultrafast shape recognition algorithm, and a planeness-check mechanism. The TGMin program has been successfully applied to search the global minima of a number of nanoclusters and periodic structures, including B30, B35, B36, B39, B40, CoB18−, RhB18−, MnB16−, and Au7 on the α-Al2O3(0 0 0 1) surface. An overview of the TGMin code and several of its recent applications are presented here.Download high-res image (164KB)Download full-size image
Co-reporter:Cong-Qiao XuMal-Soon Lee, Yang-Gang WangDavid C. Cantu, Jun LiVassiliki-Alexandra Glezakou, Roger Rousseau
ACS Nano 2017 Volume 11(Issue 2) pp:
Publication Date(Web):January 25, 2017
DOI:10.1021/acsnano.6b07409
The structure, composition, and atomic distribution of nanoalloys under operating conditions are of significant importance for their catalytic activity. In the present work, we use ab initio molecular dynamics simulations to understand the structural behavior of Au–Pd nanoalloys supported on rutile TiO2 under different conditions. We find that the Au–Pd structure is strongly dependent on the redox properties of the support, originating from strong metal–support interactions. Under reducing conditions, Pd atoms are inclined to move toward the metal/oxide interface, as indicated by a significant increase of Pd–Ti bonds. This could be attributed to the charge localization at the interface that leads to Coulomb attractions to positively charged Pd atoms. In contrast, under oxidizing conditions, Pd atoms would rather stay inside or on the exterior of the nanoparticle. Moreover, Pd atoms on the alloy surface can be stabilized by hydrogen adsorption, forming Pd–H bonds, which are stronger than Au–H bonds. Our work offers critical insights into the structure and redox properties of Au–Pd nanoalloy catalysts under working conditions.Keywords: ab initio molecular dynamics; Au−Pd nanoalloy; charge transfer; redox property; TiO2;
Co-reporter:Dr. Chaoxian Chi;Dr. Jia-Qi Wang;Hui Qu;Wan-Lu Li;Dr. Luyan Meng; Mingbiao Luo; Dr. Jun Li; Dr. Mingfei Zhou
Angewandte Chemie 2017 Volume 129(Issue 24) pp:7036-7040
Publication Date(Web):2017/06/06
DOI:10.1002/ange.201703525
AbstractWe report the preparation of UFe(CO)3− and OUFe(CO)3− complexes using a laser-vaporization supersonic ion source in the gas phase. These compounds were mass-selected and characterized by infrared photodissociation spectroscopy and state-of-the-art quantum chemical studies. There are unprecedented triple bonds between U 6d/5f and Fe 3d orbitals, featuring one covalent σ bond and two Fe-to-U dative π bonds in both complexes. The uranium and iron elements are found to exist in unique formal U(I or III) and Fe(−II) oxidation states, respectively. These findings suggest that there may exist a whole family of stable df–d multiple-bonded f-element-transition-metal compounds that have not been fully recognized to date.
Co-reporter:Teng-Teng Chen;Wan-Lu Li;Tian Jian;Xin Chen; Dr. Jun Li; Dr. Lai-Sheng Wang
Angewandte Chemie 2017 Volume 129(Issue 24) pp:7020-7024
Publication Date(Web):2017/06/06
DOI:10.1002/ange.201703111
AbstractThe structure and bonding of a Pr-doped boron cluster (PrB7−) are investigated using photoelectron spectroscopy and quantum chemistry. The adiabatic electron detachment energy of PrB7− is found to be low [1.47(8) eV]. A large energy gap is observed between the first and second detachment features, indicating a highly stable neutral PrB7. Global minimum searches and comparison between experiment and theory show that PrB7− has a half-sandwich structure with C6v symmetry. Chemical bonding analyses show that PrB7− can be viewed as a PrII[η7-B73−] complex with three unpaired electrons, corresponding to a Pr (4f26s1) open-shell configuration. Upon detachment of the 6s electron, the neutral PrB7 cluster is a highly stable PrIII[η7-B73−] complex with Pr in its favorite +3 oxidation state. The B73− ligand is found to be highly stable and doubly aromatic with six delocalized π and six delocalized σ electrons and should exist for a series of lanthanide MIII[η7-B73−] complexes.
Co-reporter:Teng-Teng Chen;Wan-Lu Li;Tian Jian;Xin Chen; Dr. Jun Li; Dr. Lai-Sheng Wang
Angewandte Chemie International Edition 2017 Volume 56(Issue 24) pp:6916-6920
Publication Date(Web):2017/06/06
DOI:10.1002/anie.201703111
AbstractThe structure and bonding of a Pr-doped boron cluster (PrB7−) are investigated using photoelectron spectroscopy and quantum chemistry. The adiabatic electron detachment energy of PrB7− is found to be low [1.47(8) eV]. A large energy gap is observed between the first and second detachment features, indicating a highly stable neutral PrB7. Global minimum searches and comparison between experiment and theory show that PrB7− has a half-sandwich structure with C6v symmetry. Chemical bonding analyses show that PrB7− can be viewed as a PrII[η7-B73−] complex with three unpaired electrons, corresponding to a Pr (4f26s1) open-shell configuration. Upon detachment of the 6s electron, the neutral PrB7 cluster is a highly stable PrIII[η7-B73−] complex with Pr in its favorite +3 oxidation state. The B73− ligand is found to be highly stable and doubly aromatic with six delocalized π and six delocalized σ electrons and should exist for a series of lanthanide MIII[η7-B73−] complexes.
Co-reporter:Dr. Chaoxian Chi;Dr. Jia-Qi Wang;Hui Qu;Wan-Lu Li;Dr. Luyan Meng; Mingbiao Luo; Dr. Jun Li; Dr. Mingfei Zhou
Angewandte Chemie International Edition 2017 Volume 56(Issue 24) pp:6932-6936
Publication Date(Web):2017/06/06
DOI:10.1002/anie.201703525
AbstractWe report the preparation of UFe(CO)3− and OUFe(CO)3− complexes using a laser-vaporization supersonic ion source in the gas phase. These compounds were mass-selected and characterized by infrared photodissociation spectroscopy and state-of-the-art quantum chemical studies. There are unprecedented triple bonds between U 6d/5f and Fe 3d orbitals, featuring one covalent σ bond and two Fe-to-U dative π bonds in both complexes. The uranium and iron elements are found to exist in unique formal U(I or III) and Fe(−II) oxidation states, respectively. These findings suggest that there may exist a whole family of stable df–d multiple-bonded f-element-transition-metal compounds that have not been fully recognized to date.
Co-reporter:Wan-Lu Li, Hong-Tao Liu, Tian Jian, Gary V. Lopez, Zachary A. Piazza, Dao-Ling Huang, Teng-Teng Chen, Jing Su, Ping Yang, Xin Chen, Lai-Sheng Wang and Jun Li
Chemical Science 2016 vol. 7(Issue 1) pp:475-481
Publication Date(Web):13 Oct 2015
DOI:10.1039/C5SC03568F
We report a joint photoelectron spectroscopy and theoretical investigation of the gaseous Au2I3− cluster, which is found to exhibit two types of isomers due to competition between Au–I covalent bonding and Au–Au aurophilic interactions. The covalent bonding favors a bent IAuIAuI− structure with an obtuse Au–I–Au angle (100.7°), while aurophilic interactions pull the two Au atoms much closer, leading to an acutely bent structure (72.0°) with an Au–Au distance of 3.08 Å. The two isomers are separated by a small barrier and are nearly degenerate with the obtuse isomer being slightly more stable. At low temperature, only the obtuse isomer is observed; distinct experimental evidence is observed for the co-existence of a combination of isomers with both acute and obtuse bending angles at room temperature. The two bond-bending isomers of Au2I3− reveal a unique example of one molecule being able to oscillate between different structures as a result of two competing chemical forces.
Co-reporter:Jin-Cheng Liu, Yan Tang, Chun-Ran Chang, Yang-Gang Wang, and Jun Li
ACS Catalysis 2016 Volume 6(Issue 4) pp:2525
Publication Date(Web):March 7, 2016
DOI:10.1021/acscatal.6b00021
We have studied the mechanism of propene epoxidation with O2–H2O mixture on Au7/α-Al2O3(0001) using global minimum approach, density functional theory (DFT), and ab initio molecular dynamics (AIMD) methods. It is found that water can easily dissociate on coordinatively unsaturated surface Al sites to form a hydroxylated Al2O3 surface. The Au7 cluster on such a surface prefers anchoring at bare Al sites and transfers from upright to flat structures with a decrease of surface hydroxyl groups. The activation of molecular oxygen via a hydroperoxyl (OOH) intermediate (i.e., O2 abstracting a hydrogen atom from coadsorbed water) is identified to be a feasible pathway from both AIMD simulations and static DFT calculations. Additional water promotes the H-transfer process by constructing a hydrogen-bonding chain with reactants. The resulting OOH turns out to be a key oxidative species for subsequent propene epoxidation. It can either dissociate into atomically oxygen species to epoxide propene or combine with propene directly, forming an important *C3H6OOH species that could be easily transformed to epoxypropane. This study reveals the important role of water in propene epoxidation and may stimulate further exploration and the usage of O2–H2O mixture as oxidizing agent in other oxidation reactions.Keywords: AIMD; gold cluster; hydroperoxyl; O2 activation; propene epoxidation; water
Co-reporter:Wei Huang, Wen-Hua Xu, W. H. E. Schwarz, and Jun Li
Inorganic Chemistry 2016 Volume 55(Issue 9) pp:4616-4625
Publication Date(Web):April 13, 2016
DOI:10.1021/acs.inorgchem.6b00442
Metal tetraoxygen molecules (MO4, M = Fe, Ru, Os, Hs, Sm, Pu) of all metal atoms M with eight valence electrons are theoretically studied using density functional and correlated wave function approaches. The heavier d-block elements Ru, Os, Hs are confirmed to form stable tetraoxides of Td symmetry in 1A1 electronic states with empty metal d0 valence shell and closed-shell O2– ligands, while the 3d-, 4f-, and 5f-elements Fe, Sm, and Pu prefer partial occupation of their valence shells and peroxide or superoxide ligands at lower symmetry structures with various spin couplings. The different geometric and electronic structures and chemical bonding types of the six iso-stoichiometric species are explained in terms of atomic orbital energies and orbital radii. The variations found here contribute to our general understanding of the periodic trends of oxidation states across the periodic table.
Co-reporter:Wei Huang, Deng-Hui Xing, Jun-Bo Lu, Bo Long, W. H. Eugen Schwarz, and Jun Li
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 4) pp:1525-1533
Publication Date(Web):March 3, 2016
DOI:10.1021/acs.jctc.5b01040
A thorough theoretical study of the relative energies of various molecular Fe·4O isomers with different oxidation states of both Fe and O atoms is presented, comparing simple Hartree–Fock through many Kohn–Sham approximations up to extended coupled cluster and DMRG multiconfiguration benchmark methods. The ground state of Fe·4O is a singlet, hexavalent iron(VI) complex 1C2v-[Fe(VI)O2]2+(O2)2–, with isomers of oxidation states Fe(II), Fe(III), Fe(IV), Fe(V), and Fe(VIII) all lying slightly higher within the range of 1 eV. The disputed existence of oxidation state Fe(VIII) is discussed for isolated FeO4 molecules. Density functional theory (DFT) at various DF approximation (DFA) levels of local and gradient approaches, Hartree–Fock exchange and meta hybrids, range dependent, DFT–D and DFT+U models do not perform better for the relative stabilities of the geometric and electronic Fe·4O isomers than within 1–5 eV. The Fe·4O isomeric species are an excellent testing and validation ground for the development of density functional and wave function methods for strongly correlated multireference states, which do not seem to always follow chemical intuition.
Co-reporter:Fu-Xing Pan, Cong-Qiao Xu, Lei-Jiao Li, Xue Min, Jian-Qiang Wang, Jun Li, Hua-Jin Zhai and Zhong-Ming Sun
Dalton Transactions 2016 vol. 45(Issue 9) pp:3874-3879
Publication Date(Web):21 Jan 2016
DOI:10.1039/C6DT00028B
We describe here the synthesis and characterization of a ternary cluster compound [As3Nb(As3Sn3)]3− (1), in which a niobium(V) atom is coordinated by an As33− triangle and a bowl-type As3Sn35− ligand. Cluster 1 was synthesized by dissolving K8NbSnAs5 (2) in the presence of [2.2.2]crypt in ethylenediamine solution, filtered and layered with toluene, then crystallized in the form of [K([2.2.2]crypt)]3[As3Nb(As3Sn3)]·en·tol. The flower-vase shaped compound 1 features a new structure type, rather different from the known Zintl phases. The stability and bonding of 1 are elucidated via extensive bonding analyses. The Sn3 ring is found to have σ-aromaticity featuring a delocalized Sn–Sn–Sn σ bond. Electronic structure calculations confirm the Nb(V) oxidation state and weak Nb–Sn and Sn–Sn bonding, in addition to the normal Nb–As and As–As bonds.
Co-reporter:Wan-Lu Li, Cong-Qiao Xu, Shu-Xian Hu and Jun Li
Dalton Transactions 2016 vol. 45(Issue 29) pp:11657-11667
Publication Date(Web):07 Mar 2016
DOI:10.1039/C6DT00602G
Recently an all-metal aromatic sandwich compound of a [Sb3Au3Sb3]3− ion has been synthesized and characterized experimentally, which indicates that there might exist a variety of stable all-metal sandwich complexes. The intralayer and interlayer chemical bonding interaction in this system plays significant roles in their stability, chemical properties and functionalities. Here we report a systematic theoretical study on the geometries, electronic structures, and chemical bonding of the [Sb3Au3Sb3]3− ion and its congeners of [X3Au3X3]3− (X = N, P, As, Sb, Bi, Uup) as well as [X3M3X3]3− (M, X = Cu, As; Ag, Bi; Au, Sb; Rg, Uup) to understand the special stabilities of these species. Additional studies are also performed on the oligomers [Sb3(Au3Sb3)n]3− (n = 1–4) to explore whether the sandwich compound can form stable extended systems. Through extensive theoretical analyses, we have shown that among the [Au3X6]3− (X = N, P, As, Sb, Bi, Uup) species, [Sb3Au3Sb3]3− is most stable due to superb matching of Sb3 and Au3 in both geometric size and fragment orbital energies. The significant stability of the [Au3Sb6]3− ion is determined by the interlayer (p-d-p)σ interactions between the vertical Au 5d6s hybrid orbitals of Au3 and Sb 5pπ orbitals of the Sb3 rings. Each Sb3 ring demonstrates unique σ aromaticity, which remains when the complex is extended to oligomers. The results suggest that it is likely that there might exist other stable [ApMpAp]x− (M = transition metals, A = main group elements, p = 3, 4, 5, …) sandwich ions and oligomers.
Co-reporter:Cong-Qiao Xu;Xiao-Gen Xiong;Wan-Lu Li
European Journal of Inorganic Chemistry 2016 Volume 2016( Issue 9) pp:1395-1404
Publication Date(Web):
DOI:10.1002/ejic.201600002
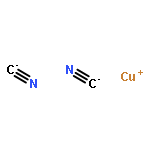
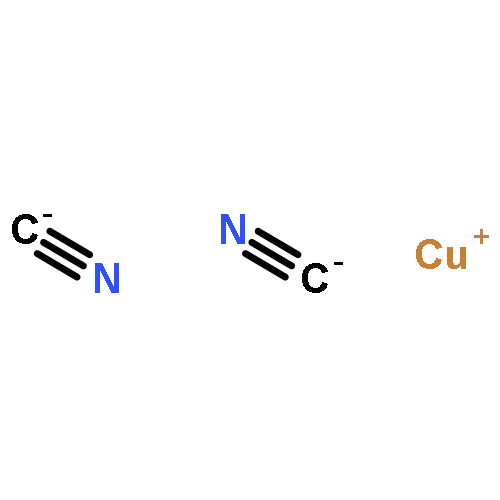
Abstract
Covalency and ionicity are key concepts in chemical science. Here we report a systematic investigation of the electronic structures and chemical bonding of [MX2]– (M = Cu, Ag, Au, Rg; X = H, Cl, CN) to illustrate the periodicity and covalency of the group 11 binary compounds. Various modern chemical bonding theories have been utilized to analyze the interactions between the metal ions and ligands. The energy decomposition approach (EDA) results suggest that the strength of the metal–ligand bonding in [MX2]– increases from Cu to Rg. Both relativistic effects and electron correlation play vital roles in the periodicity and enhancement of covalency in the heavier binary complexes. A gradual transition from Cu–X bonding with apparent ionic character to Rg–X bonding with relatively strong covalent character has been revealed from the chemical bonding analyses. The covalency of the M–X bond is enhanced by the strong relativistic effects of the metal and the decreased electronegativity of the ligands. In all the complexes studied, the monovalent metal(I) atoms interact with the neighboring atoms through significant s–d hybridizations. Spin–orbit coupling is found to be significant in [RgX2]–, and the splitting energy of the spinors is as large as 2.5 eV.
Co-reporter:Wan-Lu Li;Tian Jian;Xin Chen;Teng-Teng Chen;Dr. Gary V. Lopez;Dr. Jun Li;Dr. Lai-Sheng Wang
Angewandte Chemie International Edition 2016 Volume 55( Issue 26) pp:
Publication Date(Web):
DOI:10.1002/anie.201682661
Co-reporter:Wan-Lu Li;Tian Jian;Xin Chen;Teng-Teng Chen;Dr. Gary V. Lopez;Dr. Jun Li;Dr. Lai-Sheng Wang
Angewandte Chemie International Edition 2016 Volume 55( Issue 26) pp:7358-7363
Publication Date(Web):
DOI:10.1002/anie.201601548
Abstract
Monolayer-boron (borophene) has been predicted with various atomic arrangements consisting of a triangular boron lattice with hexagonal vacancies. Its viability was confirmed by the observation of a planar hexagonal B36 cluster with a central six-membered ring. Here we report a planar boron cluster doped with a transition-metal atom in the boron network (CoB18−), suggesting the prospect of forming stable hetero-borophenes. The CoB18− cluster was characterized by photoelectron spectroscopy and quantum chemistry calculations, showing that its most stable structure is planar with the Co atom as an integral part of a triangular boron lattice. Chemical bonding analyses show that the planar CoB18− is aromatic with ten π-electrons and the Co atom has strong covalent interactions with the surrounding boron atoms. The current result suggests that transition metals can be doped into the planes of borophenes to create metallo-borophenes, opening vast opportunities to design hetero-borophenes with tunable chemical, magnetic, and optical properties.
Co-reporter:Wan-Lu Li;Tian Jian;Xin Chen;Teng-Teng Chen;Dr. Gary V. Lopez;Dr. Jun Li;Dr. Lai-Sheng Wang
Angewandte Chemie 2016 Volume 128( Issue 26) pp:
Publication Date(Web):
DOI:10.1002/ange.201682661
Co-reporter:Wan-Lu Li;Tian Jian;Xin Chen;Teng-Teng Chen;Dr. Gary V. Lopez;Dr. Jun Li;Dr. Lai-Sheng Wang
Angewandte Chemie 2016 Volume 128( Issue 26) pp:7484-7489
Publication Date(Web):
DOI:10.1002/ange.201601548
Abstract
Monolayer-boron (borophene) has been predicted with various atomic arrangements consisting of a triangular boron lattice with hexagonal vacancies. Its viability was confirmed by the observation of a planar hexagonal B36 cluster with a central six-membered ring. Here we report a planar boron cluster doped with a transition-metal atom in the boron network (CoB18−), suggesting the prospect of forming stable hetero-borophenes. The CoB18− cluster was characterized by photoelectron spectroscopy and quantum chemistry calculations, showing that its most stable structure is planar with the Co atom as an integral part of a triangular boron lattice. Chemical bonding analyses show that the planar CoB18− is aromatic with ten π-electrons and the Co atom has strong covalent interactions with the surrounding boron atoms. The current result suggests that transition metals can be doped into the planes of borophenes to create metallo-borophenes, opening vast opportunities to design hetero-borophenes with tunable chemical, magnetic, and optical properties.
Co-reporter:Jing Su, Wei-Li Li, Gary V. Lopez, Tian Jian, Guo-Jin Cao, Wan-Lu Li, W. H. Eugen Schwarz, Lai-Sheng Wang, and Jun Li
The Journal of Physical Chemistry A 2016 Volume 120(Issue 7) pp:1084-1096
Publication Date(Web):January 29, 2016
DOI:10.1021/acs.jpca.5b11354
Uranium oxide clusters UOx– (x = 3–5) were produced by laser vaporization and characterized by photoelectron spectroscopy and quantum theory. Photoelectron spectra were obtained for UOx– at various photon energies with well-resolved detachment transitions and vibrational resolution for x = 3 and 4. The electron affinities of UOx were measured as 1.12, 3.60, and 4.02 eV for x = 3, 4, and 5, respectively. The geometric and electronic structures of both the anions and the corresponding neutrals were investigated by quasi-relativistic electron-correlation quantum theory to interpret the photoelectron spectra and to provide insight into their chemical bonding. For UOx clusters with x ≤ 3, the O atoms appear as divalent closed-shell anions around the U atom, which is in various oxidation states from UII(fds)4 in UO to UVI(fds)0 in UO3. For x > 3, there are no longer sufficient valence electrons from the U atom to fill the O(2p) shell, resulting in fractionally charged and multicenter delocalized valence states for the O ligands as well as η1- or η2-bonded O2 units, with unusual spin couplings and complicated electron correlations in the unfilled poly oxo shell. The present work expands our understanding of both the bonding capacities of actinide elements with extended spdf valence shells as well as the multitude of oxygen’s charge and bonding states.
Co-reporter:Jing Su;Shuxian Hu;Wei Huang;Mingfei Zhou
Science China Chemistry 2016 Volume 59( Issue 4) pp:442-451
Publication Date(Web):2016 April
DOI:10.1007/s11426-015-5481-z
Relativistic quantum chemistry investigations are carried out to tackle the puzzling oxidation state problem in a series of MO3- trioxide anions of all d- and f-block elements with five valence electrons. We have shown here that while the oxidation states of V, Nb, Ta, Db, Pa are, as usual, all +V with divalent oxygen O(-II) in MO3- anions, the lanthanide elements Pr and Gd cannot adopt such high +V oxidation state in similar trioxide anions. Instead, lanthanide element Gd retains its usual +III oxidation state, while Pr retains a +IV oxidation state, thus forcing oxygen into a non-innocent ligand with an uncommon monovalent radical (O•) of oxidation state -I. A unique Pr• - •(O)3 biradical with highly delocalized unpairing electron density on Pr(IV) and three O atoms is found to be responsible for stabilizing the monovalent-oxygen species in PrO3- ion, while GdO3- ion is in fact an OGd+(O22- ) complex with Gd(III). These results show that a naïve assignment of oxidation state of a chemical element without electronic structure analysis can lead to erroneous conclusions.
Co-reporter:Yan Tang, Shu Zhao, Bo Long, Jin-Cheng Liu, and Jun Li
The Journal of Physical Chemistry C 2016 Volume 120(Issue 31) pp:17514-17526
Publication Date(Web):July 15, 2016
DOI:10.1021/acs.jpcc.6b05338
Fundamental understanding of support effects and metal–support interaction is critical in heterogeneous catalysis. In this work, theoretical investigations are carried out to study the nature of support effects of different tetravalent-metal dioxides of MO2 (M = Ti, Zr, Ce, Hf, Th) in single-atom gold catalysts using density functional theory with on-site Coulomb interactions (DFT+U). The properties of gold adatom on the stoichiometric (MO2) and reduced (MO2–x) surfaces as well as CO adsorption on Au1/MO2 and Au1/MO2–x have been investigated systematically. Our calculations indicate that the fundamental quantum primogenic effect that causes the radial contraction and low orbital energies of 3d and 4f orbitals in these MO2 oxides plays a vital role in determining the valence states and charge distribution of single-atom gold as well as the adsorption modes of CO on various MO2 supports. We find that gold atoms supported on different surfaces exhibit oxidation states from Au(−I) to Au(0) to Au(I), depending on the nature of the metal oxide supports. The support-dependent oxidation states and charge distribution of Au can further influence the adsorption mode of CO. While CO adsorbs strongly on Au(I) in Au1/MO2 (M = Ti, Ce) via Au–C σ-bonding, weaker adsorption occurs on Au(0) in Au1/MO2 (M = Zr, Hf, Th) with charge back-donation to CO 2π* antibonding orbitals. In contrast, at Au1/MO2–x of reduced support, CO adsorption is stronger for M = Zr, Hf, and Th than for M = Ce. These results provide essential understanding on the nature of support effects of metal oxides in heterogeneous catalysis.
Co-reporter:Botao Qiao, Jiaxin Liu, Yang-Gang Wang, Qingquan Lin, Xiaoyan Liu, Aiqin Wang, Jun Li, Tao Zhang, and Jingyue (Jimmy) Liu
ACS Catalysis 2015 Volume 5(Issue 11) pp:6249
Publication Date(Web):September 17, 2015
DOI:10.1021/acscatal.5b01114
Preferential oxidation of CO (PROX) in H2-rich stream is critical to the production of clean H2 for the H2-based fuel cells, which provide clean and efficient energy conversion. Development of highly active and selective PROX catalysts is highly desirable but proved to be extremely challenging. Here we report that CeO2-supported Au single atoms (Au1/CeO2) are highly active, selective, and extremely stable for PROX at the PEMFC working temperature (∼80 °C) with >99.5% CO conversion over a wide temperature window, 70–120 °C (or 50–100 °C, depending on the Au loading). The high CO conversion realized at high temperatures is attributed to the unique property of single-atom catalysts that is unable to dissociatively adsorb H2 and thus has a low reactivity toward H2 oxidation. This strategy is proven in general and can be extended to other oxide-supported Au atoms (e.g., Au1/FeOx), which may open a new window for the efficient catalysis of the PROX reaction.Keywords: carbon monoxide; electron microscopy; gold; preferential oxidation; single-atom catalysis
Co-reporter:Ning Jiang; W. H. Eugen Schwarz
Inorganic Chemistry 2015 Volume 54(Issue 15) pp:7171-7180
Publication Date(Web):July 10, 2015
DOI:10.1021/acs.inorgchem.5b00372
We here report a systematic theoretical study on geometries, electronic structures, and energetic stabilities of six hexanuclear polyoxometalates [M6O19]2– of the six-valence-electron metals including the d-elements M = Cr, Mo, W, Sg from group 6 and the f-elements M = Nd, U. Scalar relativistic density functional theory was applied to these clusters in vacuum and in solution. It is shown that the Oh Lindqvist structure of the isolated [M6O19]2– units with hexavalent M elements (M+6) is only stable for the three heavy transition metals M = Mo, W, and Sg. The rare Th symmetry is predicted for M = U both in vacuum and in solution, owing to pseudo-Jahn–Teller distortion of these closed-shell systems. The Oh and Th structures correspond to cyclic “aromatic” U–̇O–̇U and alternating U=O–U bonding of cross-linked U4O4 rings, respectively. The reduced [U6O19]8– cluster with pentavalent U+5 also shows Th symmetry in vacuum, but Oh symmetry in a dielectric environment. The occurrence of different structures for varying fractional oxidation states in different environments is rationalized. Theoretical investigation of the recently synthesized U+5 complex [U6O13L6]0 (L6 = tetracyclopentadienyl dibipyridine) shows a distorted Th-type symmetry, too. The stabilities of these complexes of different metal oxidation states are consistent with the general periodic trends of oxidation states.
Co-reporter:Guo-Jin Cao; W. H. Eugen Schwarz
Inorganic Chemistry 2015 Volume 54(Issue 7) pp:3695-3701
Publication Date(Web):March 23, 2015
DOI:10.1021/acs.inorgchem.5b00356
M@Au12 cage molecules (M = transition element from group 6) are interesting clusters with high-symmetric structure and significant stability. As the heavier homologue of W is 106Sg, it is interesting to pinpoint whether the Sg@Au12 cluster is also stable. Geometric and electronic structures and bonding of various Sg@Au12 isomers were investigated with density functional theory (PW91, PBE, B3LYP) and wave function theory (MP2, CCSD(T)) approaches. The lowest-energy isomer of Sg@Au12 has icosahedral symmetry with significant Sg(6d)–Au(6s) covalent-metallic interaction and is comparable to the lighter homologues (M = Mo, W), with similar binding energy, although Sg follows (as a rare case) the textbook rule “ns below (n – 1)d”. The 12 6s valence electrons from Au12 and the six 7s6d ones from Sg can be viewed as an 18e system below and above the interacting Au 5d band, forming nine delocalized multicenter bond pairs with a high stability of ∼0.8 eV of bond energy per each of the 12 Sg–Au contacts. Different prescriptions (orbital, multipole-deformation, charge-partition, and X-ray-spectroscopy based ones) assign ambiguous atomic charges to the centric and peripheral atoms; atomic core-level energy shifts correspond to some negative charge shift to the gold periphery, more so for Cr@Au12 than for Sg@Au12 or Au@Au12.
Co-reporter:Wan-Lu Li, Yong Li, Cong-Qiao Xu, Xue-Bin Wang, Erich Vorpagel, and Jun Li
Inorganic Chemistry 2015 Volume 54(Issue 23) pp:11157-11167
Publication Date(Web):November 9, 2015
DOI:10.1021/acs.inorgchem.5b01489
Systematic theoretical and experimental investigations have been performed to understand the periodicity, electronic structures, and bonding of gold halides using tetrahalide [AuX4]− anions (X = F, Cl, Br, I, At, Uus). The [AuX4]− (X = Cl, Br, I) anions were experimentally produced in the gas phase, and their negative-ion photoelectron spectra were obtained, exhibiting rich and well-resolved spectral peaks. As expected, Au–X bonds in such series contain generally increasing covalency when halogen ligands become heavier. We calculated the adiabatic electron detachment energies as well as vertical electron detachment energies using density functional theory methods with scalar relativistic and spin–orbit coupling effects. The computationally simulated photoelectron spectra are in good agreement with the experimental ones. Our results show that the trivalent AuIII oxidation state becomes progressively less stable while AuI tends to be preferred when the halides become heavier along the Periodic Table. This series of molecules provides an example for manipulating the oxidation state of metals in complexes through ligand design.
Co-reporter:John K. Gibson, Han-Shi Hu, Michael J. Van Stipdonk, Giel Berden, Jos Oomens, and Jun Li
The Journal of Physical Chemistry A 2015 Volume 119(Issue 14) pp:3366-3374
Publication Date(Web):March 18, 2015
DOI:10.1021/jp512599e
The gas-phase complex UO2(TMOGA)22+ (TMOGA = tetramethyl-3-oxa-glutaramide) prepared by electrospray ionization was characterized by infrared multiphoton dissociation (IRMPD) spectroscopy. The IRMPD spectrum from 700–1800 cm–1 was interpreted using a computational study based on density functional theory. The predicted vibrational frequencies are in good agreement with the measured values, with an average deviation of only 8 cm–1 (<1%) and a maximum deviation of 21 cm–1 (<2%). The only IR peak assigned to the linear uranyl moiety was the asymmetric ν3 mode, which appeared at 965 cm–1 and was predicted by DFT as 953 cm–1. This ν3 frequency is red-shifted relative to bare uranyl, UO22+, by ca. 150 cm–1 due to electron donation from the TMOGA ligands. Based on the degree of red-shifting, it is inferred that two TMOGA oxygen-donor ligands have a greater effective gas basicity than the four monodentate acetone ligands in UO2(acetone)42+. The uranyl ν3 frequency was also computed for uranyl coordinated by two TMGA ligands, in which the central Oether of TMOGA has been replaced by CH2. The computed ν3 for UO2(TMGA)22+, 950 cm–1, is essentially the same as that for UO2(TMOGA)22+, suggesting that electron donation to uranyl from the Oether of TMOGA is minor. The computed ν3 asymmetric stretching frequencies for the three actinyl complexes, UO2(TMOGA)22+, NpO2(TMOGA)22+ and PuO2(TMOGA)22+, are comparable. This similarity is discussed in the context of the relationship between ν3 and intrinsic actinide-oxygen bond energies in actinyl complexes.
Co-reporter:Qiang Chen, Wei-Li Li, Ya-Fan Zhao, Su-Yan Zhang, Han-Shi Hu, Hui Bai, Hai-Ru Li, Wen-Juan Tian, Hai-Gang Lu, Hua-Jin Zhai, Si-Dian Li, Jun Li, and Lai-Sheng Wang
ACS Nano 2015 Volume 9(Issue 1) pp:754
Publication Date(Web):December 17, 2014
DOI:10.1021/nn506262c
Chirality plays an important role in chemistry, biology, and materials science. The recent discovery of the B40–/0 borospherenes marks the onset of a class of boron-based nanostructures. Here we report the observation of axially chiral borospherene in the B39– nanocluster on the bases of photoelectron spectroscopy, global minimum searches, and electronic structure calculations. Extensive structural searches in combination with density functional and CCSD(T) calculations show that B39– has a C3 cage global minimum with a close-lying C2 cage isomer. Both the C3 and C2 B39– cages are chiral with degenerate enantiomers. The C3 global minimum consists of three hexagons and three heptagons around the vertical C3 axis. The C2 isomer is built on two hexagons on the top and at the bottom of the cage with four heptagons around the waist. Both the C3 and C2 axially chiral isomers of B39– are present in the experiment and contribute to the observed photoelectron spectrum. The chiral borospherenes also exhibit three-dimensional aromaticity, featuring σ and π double delocalization for all valence electrons. Molecular dynamics simulations reveal that these chiral B39– cages are structurally fluxional above room temperature, compared to the highly robust D2d B40 borospherene. The current findings add chiral members to the borospherene family and indicate the structural diversity of boron-based nanomaterials.Keywords: all-boron fullerene; axial chirality; borospherene; global minimum searches; photoelectron spectroscopy; σ and π double delocalization;
Co-reporter:Chun-Ran Chang
The Journal of Physical Chemistry C 2015 Volume 119(Issue 28) pp:16072-16081
Publication Date(Web):June 19, 2015
DOI:10.1021/acs.jpcc.5b03965
The selective oxidation of methanol to formaldehyde with molecular O2 on Au–Pd alloy surfaces has been studied by using density functional theory (DFT). We show that the existence of Pd remarkably improves the adsorption and activation of O2 on Au–Pd surfaces. In particular, the second-neighbor Pd monomer pair (Pd–SNMP) surrounded by gold atoms can provide unique active sites for the coadsorption of O2···CH3OH, thus facilitating the activation of O2 via a hydroperoxyl radical (*OOH). With the involvement of *OOH and its decomposed fragments (*O and *OH), the oxidative dehydrogenation of methanol to formaldehyde is facilely achieved on bimetallic Au–Pd surfaces, the barriers of which are calculated to be 0.02–0.45 eV on Au2Pd/(111) and AuPd/(100) surfaces. Importantly, we find that the unusual activation of O2 via an OOH pathway instead of direct dissociation on Au–Pd catalysts is mainly responsible for the enhanced activity and selectivity in the selective oxidation of alcohols. This hydroperoxyl-based mechanism reveals the intrinsic synergy of Au–Pd bimetallic catalysts in the selective oxidation of alcohols and may provide insights for designing better bimetallic catalysts.
Co-reporter:You Yu, Mingze Zhou, Franziska Kirsch, Congqiao Xu, Li Zhang, Yu Wang, Zheng Jiang, Na Wang, Jun Li, Thomas Eitinger and Maojun Yang
Cell Research 2014 24(3) pp:267-277
Publication Date(Web):December 24, 2013
DOI:10.1038/cr.2013.172
The energy-coupling factor (ECF) transporters are multi-subunit protein complexes that mediate uptake of transition-metal ions and vitamins in about 50% of the prokaryotes, including bacteria and archaea. Biological and structural studies have been focused on ECF transporters for vitamins, but the molecular mechanism by which ECF systems transport metal ions from the environment remains unknown. Here we report the first crystal structure of a NikM, TtNikM2, the substrate-binding component (S component) of an ECF-type nickel transporter from Thermoanaerobacter tengcongensis. In contrast to the structures of the vitamin-specific S proteins with six transmembrane segments (TSs), TtNikM2 possesses an additional TS at its N-terminal region, resulting in an extracellular N-terminus. The highly conserved N-terminal loop inserts into the center of TtNikM2 and occludes a region corresponding to the substrate-binding sites of the vitamin-specific S components. Nickel binds to NikM via its coordination to four nitrogen atoms, which are derived from Met1, His2 and His67 residues. These nitrogen atoms form an approximately square-planar geometry, similar to that of the metal ion-binding sites in the amino-terminal Cu2+- and Ni2+-binding (ATCUN) motif. Replacements of residues in NikM contributing to nickel coordination compromised the Ni-transport activity. Furthermore, systematic quantum chemical investigation indicated that this geometry enables NikM to also selectively recognize Co2+. Indeed, the structure of TtNikM2 containing a bound Co2+ ion has almost no conformational change compared to the structure that contains a nickel ion. Together, our data reveal an evolutionarily conserved mechanism underlying the metal selectivity of EcfS proteins, and provide insights into the ion-translocation process mediated by ECF transporters.
Co-reporter:Han-Shi Hu ; Fan Wei ; Xuefeng Wang ; Lester Andrews
Journal of the American Chemical Society 2014 Volume 136(Issue 4) pp:1427-1437
Publication Date(Web):January 2, 2014
DOI:10.1021/ja409527u
We report a series of Si(μ-X)AnF3 (An = Th, U; X = H, F) complexes with silicon–actinide(IV) single bonds and unexpected multiradical features that form rare triplet silylenes. These bridged molecules have been prepared in microscopic scale through reactions of laser-ablated uranium and thorium atoms with silicon fluorides and identified from infrared spectra in argon and neon matrixes and relativistic quantum chemical calculations. Similar neon matrix experiments for the reactions of uranium with CF4 and CHF3 were carried out for comparison. Our density functional theory calculations show that the Si–U single-bonded species Si(μ-X)UF3 (X = H, F) with U(IV) oxidation state and the quasi-agostic bridge ligand of H or F are most stable among all the isomers, whereas the naively anticipated triple-bonded species XSi≡UF3 with U(VI) oxidation state and the double-bonded species XSi•═•UF3 with U(V) oxidation state lie markedly higher in energy. Similar thorium products from reactions with XSiF3 are also found to prefer the Si(μ-X)ThF3 structures with Si–Th single bonds and bridged H or F ligands. High level ab initio wave function theory calculations with the CCSD(T) and CASPT2 methods confirm that the ground states are quintet for Si(μ-X)UF3 and triplet for Si(μ-X)ThF3 with two unpaired electrons on the silylene group. These silicon-bearing molecules as the lowest-energy isomer of XSiAnF3 represent the first silicon–actinide systems with unusual “triplet” silylenes and Si–An single bonds with multiradical character. They are in dramatic contrast to the uranium–carbon analogs, XC≡UF3, which form triple-bonded singlet ground states with C3v symmetry. The calculated vibrational frequencies of the Si(μ-X)AnF3 complexes agree well with experimental observations. These results accentuate the critical difference of chemical bonding of 3p- and 2p-row main-group elements with actinides. The Lewis electron-pair model and the octet rule break down for these silicon compounds.
Co-reporter:Wei-Li Li ; Qiang Chen ; Wen-Juan Tian ; Hui Bai ; Ya-Fan Zhao ; Han-Shi Hu ; Jun Li ; Hua-Jin Zhai ; Si-Dian Li ;Lai-Sheng Wang
Journal of the American Chemical Society 2014 Volume 136(Issue 35) pp:12257-12260
Publication Date(Web):August 20, 2014
DOI:10.1021/ja507235s
Elemental boron is electron-deficient and cannot form graphene-like structures. Instead, triangular boron lattices with hexagonal vacancies have been predicted to be stable. A recent experimental and computational study showed that the B36 cluster has a planar C6v structure with a central hexagonal hole, providing the first experimental evidence for the viability of atom-thin boron sheets with hexagonal vacancies, dubbed borophene. Here we report a boron cluster with a double-hexagonal vacancy as a new and more flexible structural motif for borophene. Photoelectron spectrum of B35– displays a simple pattern with certain similarity to that of B36–. Global minimum searches find that both B35– and B35 possess planar hexagonal structures, similar to that of B36, except a missing interior B atom that creates a double-hexagonal vacancy. The closed-shell B35– is found to exhibit triple π aromaticity with 11 delocalized π bonds, analogous to benzo(g,h,i)perylene (C22H12). The B35 cluster can be used to build atom-thin boron sheets with various hexagonal hole densities, providing further experimental evidence for the viability of borophene.
Co-reporter:Jing Su, Zheming Wang, Duoqiang Pan, and Jun Li
Inorganic Chemistry 2014 Volume 53(Issue 14) pp:7340-7350
Publication Date(Web):June 26, 2014
DOI:10.1021/ic5006852
The electronic absorption and emission spectra of free UO2F2 and its water solvated complexes below 32 000 cm–1 are investigated at the levels of ab initio CASPT2 and CCSD(T) with inclusion of scalar relativistic and spin–orbit coupling effects. The influence of the water coordination on the electronic spectra of UO2F2 is explored by investigating the excited states of solvated complexes (H2O)nUO2F2 (n = 1–3). In these uranyl complexes, water coordination is found to have appreciable influence on the 3Δ (Ω = 1g) character of the luminescent state and on the electronic spectral shape. The simulated luminescence spectral curves based on the calculated spectral parameters of (H2O)nUO2F2 from CCSD(T) approach agree well with experimental spectra in aqueous solution at both near-liquid-helium temperature and room temperature. The possible luminescence spectra of free UO2F2 in gas phase are predicted on the basis of CASPT2 and CCSD(T) results, respectively, by considering three symmetric vibration modes. The effect of competition between spin–orbit coupling and ligand field repulsion on the luminescent state properties is discussed.
Co-reporter:YiHeng Qiu;HanShi Hu;Guo Chen
Science China Chemistry 2014 Volume 57( Issue 3) pp:426-434
Publication Date(Web):2014 March
DOI:10.1007/s11426-013-5000-z
Despite its four valence electrons, carbon can at most form triple bond in ordinary organic complexes. Quadruple bonds for carbon had been considered as impossible for a long time. Recently we showed that quadruple bonding is viable in a triatomic uranium carbide oxide molecule CUO, where the terminal C is quadruply bonded with U via its nearly unhybridized 2s- and 2p-orbitals. Here we extend this new concept to a series of diatomic molecules consisting of tetravalent p-, d-, and f-elements and terminal carbide. Investigation has been focused on a series of CM-type molecules with possible quadruply-bonded carbon (QBC), CB−, CTi, CZr, CHf, CV+, CNb+, CTa+, and isoelectronic species of CUO. We have performed natural bond orbital (NBO), natural resonance theory (NRT), and atom-in-molecule (AIM) analyses at both density functional theory (DFT) and ab initio CASSCF levels to provide evidence for the feasibility of carbon quadruple bond in these systems. Our calculation results show that the C-M bond orders in these QBC species are comparable to that in CUO, indicating terminal carbides can have novel quadruple bonding when appropriate orbitals are available in the adjacent atoms.
Co-reporter:Wei-Li Li;Dr. Ya-Fan Zhao;Dr. Han-Shi Hu;Dr. Jun Li;Dr. Lai-Sheng Wang
Angewandte Chemie International Edition 2014 Volume 53( Issue 22) pp:
Publication Date(Web):
DOI:10.1002/anie.201482271
Co-reporter:Wei-Li Li;Dr. Ya-Fan Zhao;Dr. Han-Shi Hu;Dr. Jun Li;Dr. Lai-Sheng Wang
Angewandte Chemie International Edition 2014 Volume 53( Issue 22) pp:5540-5545
Publication Date(Web):
DOI:10.1002/anie.201402488
Abstract
Chirality is vital in chemistry. Its importance in atomic clusters has been recognized since the discovery of the first chiral fullerene, the D2 symmetric C76.1 A number of gold clusters have been found to be chiral,2 raising the possibility to use them as asymmetric catalysts. The discovery of clusters with enantiomeric structures is essential to design new chiral materials with tailored chemical and physical properties.3 Herein we report the first inherently chiral boron cluster of [B30]− in a joint photoelectron spectroscopy and theoretical study. The most stable structure of [B30]− is found to be quasiplanar with a hexagonal hole. Interestingly, a pair of enantiomers arising from different positions of the hexagonal hole are found to be degenerate in our global minimum searches and both should co-exist experimentally because they have identical electronic structures and give rise to identical simulated photoelectron spectra.
Co-reporter:Wei-Li Li;Dr. Ya-Fan Zhao;Dr. Han-Shi Hu;Dr. Jun Li;Dr. Lai-Sheng Wang
Angewandte Chemie 2014 Volume 126( Issue 22) pp:
Publication Date(Web):
DOI:10.1002/ange.201482271
Co-reporter:Wei-Li Li;Dr. Ya-Fan Zhao;Dr. Han-Shi Hu;Dr. Jun Li;Dr. Lai-Sheng Wang
Angewandte Chemie 2014 Volume 126( Issue 22) pp:5646-5651
Publication Date(Web):
DOI:10.1002/ange.201402488
Abstract
Chirality is vital in chemistry. Its importance in atomic clusters has been recognized since the discovery of the first chiral fullerene, the D2 symmetric C76.1 A number of gold clusters have been found to be chiral,2 raising the possibility to use them as asymmetric catalysts. The discovery of clusters with enantiomeric structures is essential to design new chiral materials with tailored chemical and physical properties.3 Herein we report the first inherently chiral boron cluster of [B30]− in a joint photoelectron spectroscopy and theoretical study. The most stable structure of [B30]− is found to be quasiplanar with a hexagonal hole. Interestingly, a pair of enantiomers arising from different positions of the hexagonal hole are found to be degenerate in our global minimum searches and both should co-exist experimentally because they have identical electronic structures and give rise to identical simulated photoelectron spectra.
Co-reporter:Jian-Biao Liu, Xin Chen, Yi-Heng Qiu, Chao-Fei Xu, W. H. Eugen Schwarz, and Jun Li
The Journal of Physical Chemistry B 2014 Volume 118(Issue 48) pp:13954-13962
Publication Date(Web):October 31, 2014
DOI:10.1021/jp509425p
LiF–ThF4 molten salt (MS) is the fuel for advanced MS reactors. Knowledge of the microscopic MS structure and dynamics is required for an understanding of the macroscopic physical and chemical properties of the MS phases. We have performed molecular dynamics simulations on LiF–ThF4 MS at different molar percentages (LiF/ThF4 = 20.0 to 0.5) and temperatures (1100 to 1400 K). Experimental deductions and recent theoretical results on the coordination structures and transport properties of the MS are well reproduced. The density of states of the [ThF8]4– species and the character of the Th–F bonding are investigated. The interplay between the microscopic structures and the dynamical properties is elucidated. Corresponding to the smaller effective radius of Zr, the activation barrier of the M4+–F– dissociation and the lifetime of the first coordination shell of M4+ are both smaller for M = Th than for M = Zr in the respective LiF–MF4 systems. The shorter Zr–F bond is stronger than the longer Th–F bond, while the coordination number of the predominant [ZrF7]3– species is smaller than that of the dominant [ThF8]4– species. An approximate formula is proposed for the lifetime of F– ions in the first solvation shell of molten MFn (M = Y, Zr, Th) in terms of the radial distribution function.
Co-reporter:Jin-Xia Liang ; Jian Lin ; Xiao-Feng Yang ; Ai-Qin Wang ; Bo-Tao Qiao ; Jingyue Liu ; Tao Zhang
The Journal of Physical Chemistry C 2014 Volume 118(Issue 38) pp:21945-21951
Publication Date(Web):September 4, 2014
DOI:10.1021/jp503769d
Through periodic density functional theory (DFT) calculations we have investigated the catalytic mechanism of CO oxidation on an Ir1/FeOx single-atom catalyst (SAC). The rate-determining step in the catalytic cycle of CO oxidation is shown to be the formation of the second CO2 between the adsorbed CO on the surface of Ir1/FeOx and the dissociated O atom from gas phase. Comparing with Pt1/FeOx catalyst, the reaction activation barrier for CO oxidation is higher by 0.62 eV and the adsorption energy for CO molecule is larger by 0.69 eV on Ir1/FeOx. These results reveal that Ir1/FeOx catalyst has a lower activity for CO oxidation than Pt1/FeOx, which is consistent with our experimental results. The results can help to understand the fundamental mechanism of monodispersed surface atoms and to design highly active single-atom catalysts.
Co-reporter:Xiao-Feng Yang, Aiqin Wang, Botao Qiao, Jun Li, Jingyue Liu, and Tao Zhang
Accounts of Chemical Research 2013 Volume 46(Issue 8) pp:1740
Publication Date(Web):July 1, 2013
DOI:10.1021/ar300361m
Supported metal nanostructures are the most widely used type of heterogeneous catalyst in industrial processes. The size of metal particles is a key factor in determining the performance of such catalysts. In particular, because low-coordinated metal atoms often function as the catalytically active sites, the specific activity per metal atom usually increases with decreasing size of the metal particles. However, the surface free energy of metals increases significantly with decreasing particle size, promoting aggregation of small clusters. Using an appropriate support material that strongly interacts with the metal species prevents this aggregation, creating stable, finely dispersed metal clusters with a high catalytic activity, an approach industry has used for a long time. Nevertheless, practical supported metal catalysts are inhomogeneous and usually consist of a mixture of sizes from nanoparticles to subnanometer clusters. Such heterogeneity not only reduces the metal atom efficiency but also frequently leads to undesired side reactions. It also makes it extremely difficult, if not impossible, to uniquely identify and control the active sites of interest.The ultimate small-size limit for metal particles is the single-atom catalyst (SAC), which contains isolated metal atoms singly dispersed on supports. SACs maximize the efficiency of metal atom use, which is particularly important for supported noble metal catalysts. Moreover, with well-defined and uniform single-atom dispersion, SACs offer great potential for achieving high activity and selectivity.In this Account, we highlight recent advances in preparation, characterization, and catalytic performance of SACs, with a focus on single atoms anchored to metal oxides, metal surfaces, and graphene. We discuss experimental and theoretical studies for a variety of reactions, including oxidation, water gas shift, and hydrogenation. We describe advances in understanding the spatial arrangements and electronic properties of single atoms, as well as their interactions with the support. Single metal atoms on support surfaces provide a unique opportunity to tune active sites and optimize the activity, selectivity, and stability of heterogeneous catalysts, offering the potential for applications in a variety of industrial chemical reactions.
Co-reporter:Yang-Gang Wang ; Yeohoon Yoon ; Vassiliki-Alexandra Glezakou ; Jun Li ;Roger Rousseau
Journal of the American Chemical Society 2013 Volume 135(Issue 29) pp:10673-10683
Publication Date(Web):June 19, 2013
DOI:10.1021/ja402063v
To probe metal particle/reducible oxide interactions density functional theory based ab initio molecular dynamics studies were performed on a prototypical metal cluster (Au20) supported on reducible oxides (rutile TiO2(110)) to implicitly account for finite temperature effects and the role of excess surface charge in the metal oxide. It is found that the charge state of the Au particle is negative in a reducing chemical environment whereas in the presence of oxidizing species coadsorbed to the oxide surface the cluster obtained a net positive charge. In the context of the well-known CO oxidation reaction, charge transfer facilitates the plasticization of Au20, which allows for a strong adsorbate induced surface reconstruction upon addition of CO leading to the formation of mobile Au–CO species on the surface. The charging/discharging of the cluster during the catalytic cycle of CO oxidation enhances and controls the amount of O2 adsorbed at oxide/cluster interface and strongly influences the energetics of all redox steps in catalytic conversions. A detailed comparison of the current findings with previous studies is presented, and generalities about the role of surface–adsorbate charge transfer for metal cluster/reducible oxide interactions are discussed.
Co-reporter:Xiao-Gen Xiong, Wen-Hua Xu, Jun Li, Pekka Pyykkö
International Journal of Mass Spectrometry 2013 Volumes 354–355() pp:15-18
Publication Date(Web):15 November 2013
DOI:10.1016/j.ijms.2013.08.006
A DFT-level study on neutral or charged Aun clusters at relativistic and non-relativistic (R/NR) levels confirms the previously suggested relativistic origin of the planar structures. In addition, a number of potentially new aspects are observed: (1) The symmetries with a large relativistic contribution to the orbital interaction term, ΔERorb, are those, already spanned by the NR 6s orbitals, but now with strong 6s–5d hybridization. (2) All planar structures have an ‘off-plane σ’ (aka δ) bonding orbital. (3) Many molecular orbitals can be seen as linear combinations of the a1 orbitals of the smallest triangular units.
Co-reporter:ChaoFei Xu;Jing Su;Xiang Xu
Science China Chemistry 2013 Volume 56( Issue 11) pp:1525-1532
Publication Date(Web):2013 November
DOI:10.1007/s11426-013-4994-6
Understanding of the bonding nature of uranyl and various ligands is the key for designing robust sequestering agents for uranium extraction from seawater. In this paper thermodynamic properties related to the complexation reaction of uranyl(VI) in aqueous solution (i.e. existing in the form of UO2(H2O)52+) by several typical ligands (L) including acetate (CH3CO2−), bicarbonate (HOCO2−), carbonate (CO32−), CH3(NH2)CNO− (acetamidoximate, AO−) and glutarimidedioximate (denoted as GDO2−) have been investigated by using relativistic density functional theory (DFT). The geometries, vibrational frequencies, natural net charges, and bond orders of the formed uranyl-L complexes in aqueous solution are studied. Based on the DFT analysis we show that the binding interaction between uranyl and amidoximate ligand is the strongest among the selected complexes. The thermodynamics of the complexation reaction are examined, and the calculated results show that complexation of uranyl with amidoximate ligands is most preferred thermodynamically. Besides, reaction paths of the substitution complexation of solvated uranyl by acetate and AO− have been studied, respectively. We have obtained two minima along the reaction path of solvated uranyl with acetate, the monodentate-acetate complex and the bidentate-acetate one, while only one minimum involving monodentate-AO complex has been located for AO− ligand. Comparing the energy barriers of the two reaction paths, we find that complexation of uranyl with AO− is more difficult in kinetics, though it is more preferable in thermodynamics. These results show that theoretical studies can help to select efficient ligands with fine-tuned thermodynamic and kinetic properties for binding uranyl in seawater.
Co-reporter:Yong Li;Jing Su;Ellen Mitchell;GuoQing Zhang
Science China Chemistry 2013 Volume 56( Issue 12) pp:1671-1681
Publication Date(Web):2013 December
DOI:10.1007/s11426-013-4965-y
Atomic energy is an important part of current energy resources. Production of nuclear weapons and applications of nuclear fuels in nuclear power plants have accumulated numerous spent fuels containing 238U compounds, which are critical nuclear materials. How to reduce the nuclear wastes and to make use of the spent uranium are key scientific issues of environmental and nuclear science. We have reviewed here the physiochemical properties and photocatalytic mechanisms of homogeneous and heterogeneous uranium-containing materials. The current research efforts demonstrate that spent fuels can become promising new photocatalytic materials.
Co-reporter:Yu Gong, Han-Shi Hu, Linfeng Rao, Jun Li, and John K. Gibson
The Journal of Physical Chemistry A 2013 Volume 117(Issue 40) pp:10544-10550
Publication Date(Web):September 9, 2013
DOI:10.1021/jp4076977
Fragmentation of actinyl(VI) complexes UVIO2(L)22+, NpVIO2(L)22+, and PuVIO2(L)22+ (L = tetramethyl-3-oxa-glutaramide, TMOGA) produced by electrospray ionization was examined in the gas phase by collision induced dissociation (CID) in a quadrupole ion trap mass spectrometer. Cleavage of the C–Oether bond was observed for all three complexes, with dominant products being UVIO2(L)(L-86)+ with charge reduction, and NpVIO2(L)(L-101)2+ and PuVIO2(L)(L-101)2+ with charge conservation. The neptunyl and plutonyl complexes also exhibited substantial L+ loss to give pentavalent complexes NpVO2(L)+ and PuVO2(L)+, whereas the uranyl complex did not, consistent with the comparative An 5f-orbital energies and the AnVIO22+/AnVO2+ (An = U, Np, Pu) reduction potentials. CID of NpVO2(L)2+ and PuVO2(L)2+ was dominated by neutral ligand loss to form NpVO2(L)+ and PuVO2(L)+, which hydrated by addition of residual water in the ion trap; UVO2(L)2+ was not observed. Theoretical calculations of the structures and bonding of the AnVIO2(L)22+ complexes using density functional theory reveal that the metal centers are coordinated by six oxygen atoms from two TMOGA ligands.
Co-reporter:Jing Su ; Kai Zhang ; W. H. Eugen Schwarz
Inorganic Chemistry 2011 Volume 50(Issue 6) pp:2082-2093
Publication Date(Web):February 22, 2011
DOI:10.1021/ic200204p
Comprehensive computational modeling of coordination structures, thermodynamic stabilities, and luminescence spectra of uranyl-glycine-water complexes [UO2(Gly)naqm]2+ (Gly = glycine, aq = H2O, n = 0−2, m = 0−5) in aqueous solution has been carried out using relativistic density functional approaches. The solvent is approximated by a dielectric continuum model and additional explicit water molecules. Detailed pictures are obtained by synergic combination of experimental and theoretical data. The optimal equatorial coordination numbers of uranyl are determined to be five. The energies of several complex conformations are competitively close to each other. In non-basic solution the most probable complex forms are those with two water ligands replaced by the bidentate carboxyl groups of zwitterionic glycine. The N,O-chelation in non-basic solution is neither entropically nor enthalpically favored. The symmetric and antisymmetric stretch vibrations of the nearly linear O−U−O unit determine the luminescence features. The shapes of the vibrationally resolved experimental solution spectra are reproduced theoretically with an empirically fitted overall line-width parameter. The calculated luminescence origins correspond to thermally populated, near-degenerate groups of the lowest electronically excited states of 3Δg and 3Φg character, originating from (U−O)σu → (U-5f)δu,ϕu configurations of the linear [OUO]2+ unit. The intensity distributions of the vibrational progressions are consistent with U−O bond-length changes around 51/2 pm. The unusually high intensity of the short wavelength foot is explained by near-degeneracy of vibrationally and electronically excited states, and by intensity enhancement through the asymmetric O−U−O stretch mode. The combination of contemporary computational chemistry and experimental techniques leads to a detailed understanding of structures, thermodynamics, and luminescence of actinide compounds, including those with bioligands.
Co-reporter:Xuefeng Wang, Han-Gook Cho, Lester Andrews, Mingyang Chen, David A. Dixon, Han-Shi Hu, and Jun Li
The Journal of Physical Chemistry A 2011 Volume 115(Issue 10) pp:1913-1921
Publication Date(Web):February 18, 2011
DOI:10.1021/jp111592e
Laser-ablated lanthanide metal atoms were condensed with CH2F2 in excess argon at 6 K or neon at 4 K. New infrared absorption bands are assigned to the oxidative addition product methylene lanthanide difluorides on the basis of deuterium substitution and vibrational frequency calculations with density functional theory (DFT). Two dominant absorptions in the 500 cm−1 region are identified as lanthanide−fluoride stretching modes for this very strong infrared absorption. The predominantly lanthanide−carbon stretching modes follow a similar trend of increasing with metal size and have characteristic 30 cm−1 deuterium and 14 cm−1 13C isotopic shifts. The electronic structure calculations show that these CH2LnF2 complexes are not analogous to the simple transition and actinide metal methylidenes with metal−carbon double bonds that have been investigated previously, because the lanthanide metals (in the +2 or +3 oxidation state) do not appear to form a π-type bond with the CH2 group. The DFT and ab initio correlated molecular orbital theory calculations predict that these complexes exist as multiradicals, with a Ln−C σ bond and a single electron on C-2p weakly coupled with fx (x = 1 (Ce), 2 (Pr), 3(Nd), etc.) electrons in the adjacent Ln-4f orbitals. The Ln−C σ bond is composed of about 15% Ln-5d,6s and 85% C-sp2 hybrid orbital. The Ln orbital has predominantly 6s and 5d character with more d-character for early lanthanides and increasing amounts of s-character across the row. The Ln−F bonds are almost purely ionic. Accordingly, the argon−neon matrix shifts are large (13−16 cm−1) for the ionic Ln−F bond stretching modes and small (∼1 cm−1) for the more covalent Ln−C bond stretching modes.
Co-reporter:Rhitankar Pal, Wei Huang, Yi-Lei Wang, Han-Shi Hu, Satya Bulusu, Xiao-Gen Xiong, Jun Li, Lai-Sheng Wang, and Xiao Cheng Zeng
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 18) pp:2288-2293
Publication Date(Web):August 22, 2011
DOI:10.1021/jz201023q
CO chemisorption onto the Au7– cluster is investigated using photoelectron spectroscopy (PES) and ab initio calculations. It is found that CO binding can induce previously unreported 2D–3D–2D structural changes. The gold motif in the most stable structure of COAu7– is an intermediate between the two known stable 2D isomers of Au7–. Two minor isomers are observed in the PES of (CO)2Au7–; one is due to an unprecedented 3D Au7– species with Cs symmetry. This 3D Cs Au7 motif becomes a major isomer in (CO)3Au7–. The most stable isomers of COAu7– and (CO)2Au7– are planar with identical Au7 motifs; the stable planar isomers of (CO)3Au7– include not only the global-minimum structure of Au7– but also a planar hexagonal Au7 motif. The PES spectrum of (CO)4Au7– is markedly different, and its most stable structure consists of the global-minimum structure of Au7–, three terminal CO, and one bridging CO.Keywords: PES (photoelectron spectroscopy); SO (spin−orbit); VDE (vertical detachment energy);
Co-reporter:Chun-Ran Chang;Yang-Gang Wang
Nano Research 2011 Volume 4( Issue 1) pp:131-142
Publication Date(Web):2011 January
DOI:10.1007/s12274-010-0083-8
We report a comprehensive theoretical investigation of the catalytic reaction mechanisms of propene epoxidation on gold nanoclusters using density functional theory (DFT). We have shown that water acts as a catalytic promoter for propene epoxidation on gold catalysts. Even without reducible supports, hydroperoxyl (OOH) and hydroxyl (OH) radicals are readily formed on small-size gold clusters from co-adsorbed H2O and O2, with energy barriers as low as 4–6 kcal/mol (1 cal = 4.186 J). Propene epoxidation occurs easily through reactions between C3H6 and the weakened O-O bond of the OOH radicals on the surfaces of gold clusters.
Co-reporter:Xiao-Feng Yang, Yi-Lei Wang, Ya-Fan Zhao, Ai-Qin Wang, Tao Zhang and Jun Li
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 12) pp:3038-3043
Publication Date(Web):04 Feb 2010
DOI:10.1039/B921367H
Understanding the geometry structures of gold clusters, especially with adsorbates, is essential for designing highly active gold nanocatalysts. Here, we report a detailed theoretical study of the geometry structures of bare and CO-saturated Aun+ (n = 4–6) clusters. It is found that the chemisorption of CO molecules leads to significant geometry changes of the gold clusters from two- to three-dimensions (3D), even for clusters as small as Au4+. These gold cationic clusters exhibit characteristic coordination binding sites that have distinct electronic structures. We also find that commonly used density functional theory (DFT) methods have difficulty in accurately predicting energies of some isomers of Aun+ clusters or Aun(CO)n+ complexes, with the calculated relative energies strongly depending on the exchange–correlation functionals used. Caution must be exercised when using DFT methods as a blackbox for predicting the structures and energies of gold clusters.
Co-reporter:Yi-Lei Wang, Xue-Bin Wang, Xiao-Peng Xing, Fan Wei, Jun Li, and Lai-Sheng Wang
The Journal of Physical Chemistry A 2010 Volume 114(Issue 42) pp:11244-11251
Publication Date(Web):May 12, 2010
DOI:10.1021/jp103173d
We report a combined experimental and theoretical investigation of MI2− (M = Cs, Cu, Ag, Au) to explore the chemical bonding in the group IA and IB diiodide complexes. Both photoelectron imaging and low-temperature photoelectron spectroscopy are applied to MI2− (M = Cs, Cu, Au), yielding vibrationally resolved spectra for CuI2− and AuI2− and accurate electron affinities, 4.52 ± 0.02, 4.256 ± 0.010, and 4.226 ± 0.010 eV for CsI2, CuI2, and AuI2, respectively. Spin−orbit coupling is found to be important in all the diiodide complexes and ab initio calculations including spin−orbit effects allow quantitative assignments of the observed photoelectron spectra. A variety of chemical bonding analyses (charge population, bond order, and electron localization functions) have been carried out, revealing a gradual transition from the expected ionic behavior in CsI2− to relatively strong covalent bonding in AuI2−. Both relativistic effects and electron correlation are shown to enhance the covalency in the gold diiodide complex.
Co-reporter:Hai Xiao, Han-Shi Hu, W. H. Eugen Schwarz, and Jun Li
The Journal of Physical Chemistry A 2010 Volume 114(Issue 33) pp:8837-8844
Publication Date(Web):June 24, 2010
DOI:10.1021/jp102107n
The existence of a novel octahedral UO6 complex had been suggested by Pyykkö et al. [Pyykkö, P.; Runeberg, N.; Straka, M.; Dyall, K. G. Chem. Phys. Lett. 2000, 328, 415]. We have now investigated the stability, the geometric and electronic structures, and the vibrations of various UO6 molecules, using spin−orbit density functional and scalar-relativistic coupled-cluster approaches. We find four different (meta-)stable species, namely 3D2h-UO2(η2-O2•)2 at lowest energy, 3C2v-UO4•(η2-O2•) and 1D3-U(η2-O2)3 at medium energies, and 1Oh-UO6 at highest energy. The decay of Oh-UO6 occurs via an activated spin-flip mechanism. The UO6 species correspond to local minima on singlet and triplet energy surfaces and might be trapped in noble gas matrices. Experimentally, the four species might be identified through their vibrational spectra. Uranium is best described as coordinated by oxygen atoms in various oxidation states as oxo O2−, oxido(1) O•−, peroxido O22−, and superoxido O2•− ligands. The occurrence of monovalent oxygen is remarkable. The resulting characterization of the central ion as UVI in all four cases does not fully reflect the electronic differences, nor the “valence-activity” of the U-6p6 semicore shell.
Co-reporter:Yi-Lei Wang, Hua-Jin Zhai, Lu Xu, Jun Li and Lai-Sheng Wang
The Journal of Physical Chemistry A 2010 Volume 114(Issue 3) pp:1247-1254
Publication Date(Web):September 18, 2009
DOI:10.1021/jp903558v
We report vibrationally resolved photoelectron spectroscopy (PES) of Au2(CO)n− (n = 1−3), in combination with relativistic density functional theory (DFT) and ab initio calculations. The ground-state transition in the spectrum of Au2CO− is broad, containing vibrational structures both in the bending and in the CO stretching modes and suggesting a large structural change from Au2CO− to Au2CO. The ground-state transitions for both n = 2 and 3 display a well-resolved vibrational progression in the CO stretching mode with frequencies of 2110 ± 40 and 2160 ± 40 cm−1, respectively. The PES data show that chemisorption of the first two CO’s each induces a significant red-shift in the electron binding energies. The third CO is physisorbed, inducing only a slight increase in electron binding energies relative to Au2(CO)2−. Relativistic DFT and ab initio calculations are performed to determine the ground-state structures for Au2(CO)n− and Au2(CO)n, and the results agree well with the experiment. Au2(CO), Au2(CO)2, and Au2(CO)2− are all found to be linear, while Au2(CO)− is bent due to the Renner−Teller effect. A strong spin−orbit effect is found in Au2(CO)2− that quenches the Renner−Teller effect, keeping the linear structure for this anion. The physisorption in Au2(CO)3− is borne out in CCSD(T) calculations. However, a wide range of DFT methods tested fail to correctly predict the relative energies of the physisorbed versus chemisorbed isomers for Au2(CO)3−.
Co-reporter:Guanjun Wang;Jing Su;Yu Gong;Mingfei Zhou Dr. Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 7) pp:1302-1305
Publication Date(Web):
DOI:10.1002/anie.200906473
Co-reporter:Xiao-Feng Yang, Ai-Qin Wang, Yi-Lei Wang, Tao Zhang and Jun Li
The Journal of Physical Chemistry C 2010 Volume 114(Issue 7) pp:3131-3139
Publication Date(Web):January 28, 2010
DOI:10.1021/jp9107415
We report a combined experimental and theoretical investigation of the unusual catalytic properties of gold nanoparticles in the selective hydrogenation of 1,3-butadiene. It is found that nanogold catalysts exhibit a unique preference toward forming cis-2-butene compared to the trans isomer, and the ratio of cis/trans isomers is structure-sensitive in terms of the size of gold nanoparticles. Our density functional theory calculations show that the cis-1,3-butadiene can be preferably adsorbed at the edges and corners of gold nanoparticles as compared to the trans-1,3-butadiene. Moreover, the transition state calculations indicate that the isomerization of trans-butadiene to cis-butadiene is kinetically favorable, with rather low energy barriers. These unique properties of gold nanoparticles toward 1,3-butadiene adsorption are discussed based on orbital interaction analyses. It is found that Au-6s-based molecular orbitals prefer adsorption of cis-1,3-butadiene to the trans isomer due to spatial match. The catalytic selectivity of gold nanoparticles toward formation of cis-2-butene has been further corroborated by comparable investigations on silver and copper catalysts.
Co-reporter:Xue-Bin Wang ; Yi-Lei Wang ; Jie Yang ; Xiao-Peng Xing ; Jun Li ;Lai-Sheng Wang
Journal of the American Chemical Society 2009 Volume 131(Issue 45) pp:16368-16370
Publication Date(Web):October 27, 2009
DOI:10.1021/ja908106e
The Au(CN)2− ion is the most stable Au compound known for centuries, yet a detailed understanding of its chemical bonding is still lacking. Here we report direct experimental evidence of significant covalent bonding character in the Au−C bonds in Au(CN)2− using photoelectron spectroscopy and comparisons with its lighter congeners, Ag(CN)2− and Cu(CN)2−. Vibrational progressions in the Au−C stretching mode were observed for all detachment transitions for Au(CN)2−, in contrast to the atomic-like transitions for Cu(CN)2−, revealing the Au−C covalent bonding character. In addition, rich electronic structural information was obtained for Au(CN)2− by employing 118 nm detachment photons. Density functional theory and high-level ab initio calculations were carried out to understand the photoelectron spectra and obtain insight into the nature of the chemical bonding in the M(CN)2− complexes. Significant covalent character in the Au−C bonding due to the strong relativistic effects was revealed in Au(CN)2−, consistent with its high stability.
Co-reporter:Zhong-Ming Sun;Ya-Fan Zhao;Lai-Sheng Wang
Journal of Cluster Science 2009 Volume 20( Issue 3) pp:601-609
Publication Date(Web):2009 September
DOI:10.1007/s10876-009-0266-1
A new Zintl cluster [Ge9PdPPh3]3− has been isolated as (2,2,2-crypt)K+ salt through the reaction of K4Ge9 and Pd[PPh3]4 in ethylenediamine solutions and characterized via single-crystal X-ray crystallography. The as-prepared bimetallic [Ge9PdPPh3]3− cluster could successfully trap a nickel atom to form a trimetallic cluster [Ni@(Ge9PdPPh3)]2−. The coordination of Ge94− by PdPPh3 induces a one-electron oxidation and encapsulation of the Ni atom into the Ge93− cage leads to a further one-electron oxidation and a geometry transformation from C4v (nido) to C3v (closo).
Co-reporter:Hua-Jin Zhai ; Li-Li Pan ; Bing Dai ; Boggavarapu Kiran ; Jun Li ;Lai-Sheng Wang
The Journal of Physical Chemistry C 2008 Volume 112(Issue 31) pp:11920-11928
Publication Date(Web):July 11, 2008
DOI:10.1021/jp803161b
The interactions of CO with gold clusters are essential to understanding the catalytic mechanisms of CO oxidation on supported gold nanoparticles. Here we report a photoelectron spectroscopy and theoretical study of CO adsorption on a well-defined Au6− cluster in Au6(CO)n− (n = 4−9). Previous studies have shown that the first three CO successively bind the three apex sites of the triangular Au6−. The current work reveals that the fourth CO induces a major structural change to create more apex sites to accommodate the additional CO. Definitive spectroscopic evidence is obtained for the chemisorption saturation at Au6(CO)6−, in which Au6 has rearranged to accommodate the six CO adsorbates. The photoelectron spectra of larger clusters from Au6(CO)7− to Au6(CO)9− are observed to be almost identical to that of Au6(CO)6−, suggesting that the additional CO units are simply physisorbed onto the Au6(CO)6− core. Quasirelativistic density functional calculations are performed on both Au6(CO)n and Au6(CO)n− (n = 4−6). The theoretical results are used to interpret the experimental observations and to provide insight into the nature of CO interactions with gold clusters. The Au6 cluster is shown to be highly fluxional upon multiple CO adsorptions, stabilizing structures with more apex sites to accommodate the additional CO units. The CO-induced structural transformation is analogous to structural flexibility and mobility in heterogeneous catalysis. The observations of the propensity of CO toward apex sites and CO-induced structural changes in small gold clusters may be important for understanding the mechanisms of CO oxidation on supported gold nanoparticles.
Co-reporter:Yanying Zhao, Jing Su, Yu Gong, Jun Li and Mingfei Zhou
The Journal of Physical Chemistry A 2008 Volume 112(Issue 37) pp:8606-8611
Publication Date(Web):August 27, 2008
DOI:10.1021/jp804995d
Laser-evaporated chromium atoms are shown to insert into dioxygen to form CrO2 in solid argon. Annealing allows diffusion and reactions to form (η2-O2)2CrO2, which is characterized as [(O2−)2(CrO2)2+], a side-on bonded disuperoxo−chromium dioxide complex. The (η2-O2)2CrO2 complex further reacts with xenon atom doped in solid argon to give (η1-OO)(η2-O2)CrO2(Xe), which can be regarded as an O2 molecule weakly interacting with [(O2)2−(CrO2)2+Xe], a side-on bonded peroxo−chromium dioxide-xenon complex. The results indicate surprisingly that xenon atom induces a disproportionation reaction from superoxo to peroxo and dioxygen complex.
Co-reporter:Mingfei Zhou Dr.;Xi Jin;Yu Gong Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 16) pp:
Publication Date(Web):9 MAR 2007
DOI:10.1002/anie.200605218
Building up from scratch: The Gd2 molecule reacts with N2 in solid argon to form a homoleptic dinuclear dinitrogen complex containing a drastically activated side-on and end-on bonded N2 ligand (see scheme; Gd orange, N blue). The complex rearranges to a planar cyclic [Gd(μ-N)2Gd] isomer with a completely cleaved NN bond, which further dimerizes to form a cubic [Gd4N4] cluster, a building block for (GdN)x nanoparticles.
Co-reporter:Jun Li Dr.;Han-Shi Hu;Jonathan T. Lyon Dr.;Lester Andrews Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 47) pp:
Publication Date(Web):17 OCT 2007
DOI:10.1002/anie.200702771
Actinide goes chiral: Theoretical and experimental investigations demonstrate that actinide methylidene complexes form agostic structures with significant pyramidality at the actinide center, leading to chirality, for example in the [H2CUFCl] complex shown. A whole series of actinide methylidene complexes [H2CAnXY] (An=Th, U; X, Y=F, Cl, Br, I, H) was investigated and shown to be chiral.
Co-reporter:Mingfei Zhou Dr.;Xi Jin;Yu Gong Dr.
Angewandte Chemie 2007 Volume 119(Issue 16) pp:
Publication Date(Web):9 MAR 2007
DOI:10.1002/ange.200605218
Von Grund auf: Gd2-Moleküle reagieren mit N2 in festem Argon zu einem homoleptischen Distickstoff-Zweikernkomplex mit stark aktiviertem side-on- und end-on-gebundenem N2-Liganden (siehe Schema; Gd orange, N blau). Der Komplex lagert sich in das planare cyclische Isomer [Gd(μ-N)2Gd] um, in dem keine N-N-Bindung mehr vorliegt; diese Spezies dimerisiert zu dem würfelförmigen Cluster [Gd4N4], der als Baustein in (GdN)x-Nanopartikeln auftritt.
Co-reporter:Jun Li Dr.;Han-Shi Hu;Jonathan T. Lyon Dr.;Lester Andrews Dr.
Angewandte Chemie 2007 Volume 119(Issue 47) pp:
Publication Date(Web):17 OCT 2007
DOI:10.1002/ange.200702771
Pyramidenkräfte: Theoretische und experimentelle Studien belegen, dass Actinoid-Methylidinkomplexe agostische Strukturen mit merklich pyramidalisiertem Actinoidzentrum bilden und somit chiral sind. Eine Reihe von chiralen Actinoid-Methylidinkomplexen [H2CAnXY] (An=Th, U; X, Y=F, Cl, Br, I, H) wurde synthetisiert und charakterisiert.
Co-reporter:Jonathan T. Lyon;Lester Andrews;Han-Shi Hu
PNAS 2007 Volume 104 (Issue 48 ) pp:18919-18924
Publication Date(Web):2007-11-27
DOI:10.1073/pnas.0707035104
Chemistry of the actinide elements represents a challenging yet vital scientific frontier. Development of actinide chemistry
requires fundamental understanding of the relative roles of actinide valence-region orbitals and the nature of their chemical
bonding. We report here an experimental and theoretical investigation of the uranium methylidyne molecules X3UCH (X = F, Cl, Br), F2ClUCH, and F3UCF formed through reactions of laser-ablated uranium atoms and trihalomethanes or carbon tetrafluoride in excess argon. By
using matrix infrared spectroscopy and relativistic quantum chemistry calculations, we have shown that these actinide complexes
possess relatively strong UC triple bonds between the U 6d-5f hybrid orbitals and carbon 2s-2p orbitals. Electron-withdrawing ligands are critical in stabilizing the U(VI) oxidation state and sustaining the formation
of uranium multiple bonds. These unique UC-bearing molecules are examples of the long-sought actinide-alkylidynes. This discovery opens the door to the rational synthesis
of triple-bonded actinidecarbon compounds.
Co-reporter:Xue-Bin Wang, Yi-Lei Wang, Hin-Koon Woo, Jun Li, Guo-Shi Wu, Lai-Sheng Wang
Chemical Physics 2006 Volume 329(1–3) pp:230-238
Publication Date(Web):26 October 2006
DOI:10.1016/j.chemphys.2006.07.018
Co-reporter:Qingfeng Ge, Jian-guo Wang, Jun Li
Catalysis Today (16 May 2011) Volume 165(Issue 1) pp:
Publication Date(Web):16 May 2011
DOI:10.1016/j.cattod.2011.03.001
Co-reporter:Ping-Xia Zhang, Yang-Gang Wang, Yan-Qiang Huang, Tao Zhang, Guo-Shi Wu, Jun Li
Catalysis Today (16 May 2011) Volume 165(Issue 1) pp:80-88
Publication Date(Web):16 May 2011
DOI:10.1016/j.cattod.2011.01.012
The mechanisms of hydrazine decompositions on Ir(1 1 1) have been investigated by using slab model based on periodic density functional theory (DFT). In order to shed light on the elementary radical reaction processes of hydrazine decomposition on Ir-based catalysts, three possible reaction pathways are considered. Through computational modeling we have investigated the adsorption characteristics, geometrical structures, activation energies, and reaction mechanisms. The initial reactants, transition states, and final products of each elementary step and various likely intermediates are discussed. We have found that the main reaction channel with relatively low energy barriers is the following: the thermal decomposition of hydrazine forms two NH2 radicals, which attack an adjacent adsorbed hydrazine molecule or subsequent N2Hx (x = 1–3) species and capture the H atoms step by step, finally leading to the formation of N2 and NH3 products. We show that the rate-determining step involves NH2 interacting with a N2H species, with an energy barrier of 0.63 eV (or 14.5 kcal/mol). The overall reaction channel releases a large amount of thermal energies. The decomposition of hydrazine on Ir surfaces is therefore both thermodynamically and kinetically favorable. The other reaction channels investigated have much higher activation barriers with Ir catalysts.
Co-reporter:Wei Huang; Pekka Pyykkö
Inorganic Chemistry () pp:
Publication Date(Web):August 26, 2015
DOI:10.1021/acs.inorgchem.5b01540
While the oxidation state Pu(VIII) is shown to be less stable than Pu(V) in the PuO4 molecule, it is not clear if the more electronegative fluorine can help to stabilize Pu(VIII). Our calculations on PuOnF8–2n (n = 0–4) molecules notably confirm that PuO2F4 has both 1D4h and 5C2v minima with the oxidation states Pu(VIII) and Pu(V), respectively, with the latter having lower energy. The hybrid-DFT, CCSD(T), and CASSCF methods all give the same result. The results conform to a superoxide ligand when n ≥ 2. PuF8 in a 1Oh state can decompose to PuF6 and F2, and PuOF6 in a 1C2v state also can break down to PuF6 and 1/2 O2. The Pu(VIII) anion PuO2F5– does have a D5h minimum, which also lies above a 5C2v Pu(V) peroxide structure. However, the energy differences between the different minima are not large, indicating that metastable species with oxidation states higher than Pu(V) cannot be completely excluded.
Co-reporter:Wan-Lu Li, Hong-Tao Liu, Tian Jian, Gary V. Lopez, Zachary A. Piazza, Dao-Ling Huang, Teng-Teng Chen, Jing Su, Ping Yang, Xin Chen, Lai-Sheng Wang and Jun Li
Chemical Science (2010-Present) 2016 - vol. 7(Issue 1) pp:NaN481-481
Publication Date(Web):2015/10/13
DOI:10.1039/C5SC03568F
We report a joint photoelectron spectroscopy and theoretical investigation of the gaseous Au2I3− cluster, which is found to exhibit two types of isomers due to competition between Au–I covalent bonding and Au–Au aurophilic interactions. The covalent bonding favors a bent IAuIAuI− structure with an obtuse Au–I–Au angle (100.7°), while aurophilic interactions pull the two Au atoms much closer, leading to an acutely bent structure (72.0°) with an Au–Au distance of 3.08 Å. The two isomers are separated by a small barrier and are nearly degenerate with the obtuse isomer being slightly more stable. At low temperature, only the obtuse isomer is observed; distinct experimental evidence is observed for the co-existence of a combination of isomers with both acute and obtuse bending angles at room temperature. The two bond-bending isomers of Au2I3− reveal a unique example of one molecule being able to oscillate between different structures as a result of two competing chemical forces.
Co-reporter:Fu-Xing Pan, Cong-Qiao Xu, Lei-Jiao Li, Xue Min, Jian-Qiang Wang, Jun Li, Hua-Jin Zhai and Zhong-Ming Sun
Dalton Transactions 2016 - vol. 45(Issue 9) pp:NaN3879-3879
Publication Date(Web):2016/01/21
DOI:10.1039/C6DT00028B
We describe here the synthesis and characterization of a ternary cluster compound [As3Nb(As3Sn3)]3− (1), in which a niobium(V) atom is coordinated by an As33− triangle and a bowl-type As3Sn35− ligand. Cluster 1 was synthesized by dissolving K8NbSnAs5 (2) in the presence of [2.2.2]crypt in ethylenediamine solution, filtered and layered with toluene, then crystallized in the form of [K([2.2.2]crypt)]3[As3Nb(As3Sn3)]·en·tol. The flower-vase shaped compound 1 features a new structure type, rather different from the known Zintl phases. The stability and bonding of 1 are elucidated via extensive bonding analyses. The Sn3 ring is found to have σ-aromaticity featuring a delocalized Sn–Sn–Sn σ bond. Electronic structure calculations confirm the Nb(V) oxidation state and weak Nb–Sn and Sn–Sn bonding, in addition to the normal Nb–As and As–As bonds.
Co-reporter:Wan-Lu Li, Cong-Qiao Xu, Shu-Xian Hu and Jun Li
Dalton Transactions 2016 - vol. 45(Issue 29) pp:NaN11667-11667
Publication Date(Web):2016/03/07
DOI:10.1039/C6DT00602G
Recently an all-metal aromatic sandwich compound of a [Sb3Au3Sb3]3− ion has been synthesized and characterized experimentally, which indicates that there might exist a variety of stable all-metal sandwich complexes. The intralayer and interlayer chemical bonding interaction in this system plays significant roles in their stability, chemical properties and functionalities. Here we report a systematic theoretical study on the geometries, electronic structures, and chemical bonding of the [Sb3Au3Sb3]3− ion and its congeners of [X3Au3X3]3− (X = N, P, As, Sb, Bi, Uup) as well as [X3M3X3]3− (M, X = Cu, As; Ag, Bi; Au, Sb; Rg, Uup) to understand the special stabilities of these species. Additional studies are also performed on the oligomers [Sb3(Au3Sb3)n]3− (n = 1–4) to explore whether the sandwich compound can form stable extended systems. Through extensive theoretical analyses, we have shown that among the [Au3X6]3− (X = N, P, As, Sb, Bi, Uup) species, [Sb3Au3Sb3]3− is most stable due to superb matching of Sb3 and Au3 in both geometric size and fragment orbital energies. The significant stability of the [Au3Sb6]3− ion is determined by the interlayer (p-d-p)σ interactions between the vertical Au 5d6s hybrid orbitals of Au3 and Sb 5pπ orbitals of the Sb3 rings. Each Sb3 ring demonstrates unique σ aromaticity, which remains when the complex is extended to oligomers. The results suggest that it is likely that there might exist other stable [ApMpAp]x− (M = transition metals, A = main group elements, p = 3, 4, 5, …) sandwich ions and oligomers.
Co-reporter:Xiao-Feng Yang, Yi-Lei Wang, Ya-Fan Zhao, Ai-Qin Wang, Tao Zhang and Jun Li
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 12) pp:NaN3043-3043
Publication Date(Web):2010/02/04
DOI:10.1039/B921367H
Understanding the geometry structures of gold clusters, especially with adsorbates, is essential for designing highly active gold nanocatalysts. Here, we report a detailed theoretical study of the geometry structures of bare and CO-saturated Aun+ (n = 4–6) clusters. It is found that the chemisorption of CO molecules leads to significant geometry changes of the gold clusters from two- to three-dimensions (3D), even for clusters as small as Au4+. These gold cationic clusters exhibit characteristic coordination binding sites that have distinct electronic structures. We also find that commonly used density functional theory (DFT) methods have difficulty in accurately predicting energies of some isomers of Aun+ clusters or Aun(CO)n+ complexes, with the calculated relative energies strongly depending on the exchange–correlation functionals used. Caution must be exercised when using DFT methods as a blackbox for predicting the structures and energies of gold clusters.
Co-reporter:Jun-Bo Lu, Jiwen Jian, Wei Huang, Hailu Lin, Jun Li and Mingfei Zhou
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 45) pp:NaN31131-31131
Publication Date(Web):2016/10/19
DOI:10.1039/C6CP06753K
The experimentally known highest oxidation state of iron has been determined to be Fe(VI) so far. Here we report a combined matrix-isolation infrared spectroscopic and theoretical study of two interconvertible iron oxide anions: a dioxoiron peroxide complex [(η2-O2)FeO2]− with a C2v-structure and a tetroxide FeO4− with a D2d tetrahedral structure, which are formed by co-condensation of laser-ablated iron atoms and electrons with O2/Ar mixtures at 4 K. Quantum chemistry theoretical studies indicate that the Jahn–Teller distorted tetroxide FeO4− anion is a d1 species with hereto the highest iron formal oxidation state Fe(VII).
Co-reporter:Jin-Xia Liang, Xiao-Feng Yang, Aiqin Wang, Tao Zhang and Jun Li
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 18) pp:NaN6892-6892
Publication Date(Web):2016/06/14
DOI:10.1039/C6CY00672H
Significant progress has recently been made in single-atom catalysis involving noble metals. We report here a theoretical investigation of the catalytic mechanism of CO oxidation of a non-noble metal single-atom catalyst (SAC) Ni1/FeOx using density functional theory (DFT). The calculated results show that this new SAC Ni1/FeOx has a high catalytic activity at room temperature for CO oxidation. CO adsorption strength is a key factor in determining catalytic activity for CO oxidation. Compared with noble-metal catalysts Pt1/FeOx and Ir1/FeOx, the catalytic activity for CO oxidation of Ni1/FeOx is found to be comparable to that of Pt1/FeOx, but is considerably higher than that of Ir1/FeOx. Our theoretical prediction of this new non-noble metal Ni1/FeOx catalyst with high catalytic activity for CO oxidation at room temperature may find practical applications. The theoretical investigation provides a fundamental understanding of the catalytic mechanism of singly-dispersed surface atoms and helps to stimulate further experimental studies on highly active non-noble metal single-atom catalysts.
Co-reporter:Wan-Lu Li, Tian Jian, Xin Chen, Hai-Ru Li, Teng-Teng Chen, Xue-Mei Luo, Si-Dian Li, Jun Li and Lai-Sheng Wang
Chemical Communications 2017 - vol. 53(Issue 10) pp:NaN1590-1590
Publication Date(Web):2016/12/20
DOI:10.1039/C6CC09570D
A tubular molecular rotor B2-Ta@B18− (1) and boron drum Ta@B20− (2) with the highest coordination number of twenty in chemistry are observed via a joint photoelectron spectroscopy and first-principles theory investigation.
Co-reporter:Shu-Xian Hu, John K. Gibson, Wan-Lu Li, Michael J. Van Stipdonk, Jonathan Martens, Giel Berden, Britta Redlich, Jos Oomens and Jun Li
Chemical Communications 2016 - vol. 52(Issue 86) pp:NaN12764-12764
Publication Date(Web):2016/10/05
DOI:10.1039/C6CC07205D
Understanding of the nature and extent of chemical bonding in uranyl coordination complexes is crucial for the design of new ligands for nuclear waste separation, uranium extraction from seawater, and other applications. We report here the synthesis, infrared spectroscopic characterization, and quantum chemical studies of a molecular uranyl–di-15-crown-5 complex. The structure and bonding of this unique complex featuring a distinctive 6-fold coplanar coordination staggered sandwich structure and an unusual non-perpendicular orientation of the uranyl moiety are evaluated using density functional theory and chemical bonding analyses. The results provide fundamental understanding of the coordination interaction of uranyl with oxygen-donor ligands.
Co-reporter:Shu-Xian Hu, Jiwen Jian, Jing Su, Xuan Wu, Jun Li and Mingfei Zhou
Chemical Science (2010-Present) 2017 - vol. 8(Issue 5) pp:NaN4043-4043
Publication Date(Web):2017/03/15
DOI:10.1039/C7SC00710H
The neutral molecule NPrO and its anion NPrO− are produced via co-condensation of laser-ablated praseodymium atoms with nitric oxide in a solid neon matrix. Combined infrared spectroscopy and state-of-the-art quantum chemical calculations confirm that both species are pentavalent praseodymium nitride-oxides with linear structures that contain PrN triple bonds and PrO double bonds. Electronic structure studies show that the neutral NPrO molecule features a 4f0 electron configuration and a Pr(V) oxidation state similar to that of the isoelectronic PrO2+ ion, while its NPrO− anion possesses a 4f1 electron configuration and a Pr(IV) oxidation state. The neutral NPrO molecule is thus a rare lanthanide nitride-oxide species with a Pr(V) oxidation state, which follows the recent identification of the first Pr(V) oxidation state in the PrO2+ and PrO4 complexes (Angew. Chem. Int. Ed., 2016, 55, 6896). This finding indicates that lanthanide compounds with oxidation states of higher than +IV are richer in chemistry than previously recognized.
Co-reporter:Tian Jian, Wan-Lu Li, Xin Chen, Teng-Teng Chen, Gary V. Lopez, Jun Li and Lai-Sheng Wang
Chemical Science (2010-Present) 2016 - vol. 7(Issue 12) pp:NaN7027-7027
Publication Date(Web):2016/07/25
DOI:10.1039/C6SC02623K
Metal-doped boron clusters provide new opportunities to design nanoclusters with interesting structures and bonding. A cobalt-doped boron cluster, CoB18−, has been observed recently to be planar and can be viewed as a motif for metallo-borophenes, whereas the D9d drum isomer as a motif for metallo-boronanotubes is found to be much higher in energy. Hence, whether larger doped boron drums are possible is still an open question. Here we report that for RhB18− the drum and quasi-planar structures become much closer in energy and co-exist experimentally, revealing a competition between the metallo-boronanotube and metallo-borophene structures. Photoelectron spectroscopy of RhB18− shows a complicated spectral pattern, suggesting the presence of two isomers. Quantum chemistry studies indicate that the D9d drum isomer and a quasi-planar isomer (Cs) compete for the global minimum. The enhanced stability of the drum isomer in RhB18− is due to the less contracted Rh4d orbitals, which can have favorable interactions with the B18 drum motif. Chemical bonding analyses show that the quasi-planar isomer of RhB18− is aromatic with 10 π electrons, whereas the observed RhB18− drum cluster sets a new record for coordination number of eighteen among metal complexes. The current finding shows that the size of the boron drum can be tuned by appropriate metal dopants, suggesting that even larger boron drums with 5d, 6d transition metal, lanthanide or actinide metal atoms are possible.
Co-reporter:Jian-Biao Liu, Guo P. Chen, Wei Huang, David L. Clark, W. H. Eugen Schwarz and Jun Li
Dalton Transactions 2017 - vol. 46(Issue 8) pp:NaN2550-2550
Publication Date(Web):2017/01/26
DOI:10.1039/C6DT03953G
Actinyl-tricarbonato anions [(AnO2)(CO3)3]4− (An = U–Cm) in various environments were investigated using theoretical approaches of quantum-mechanics, molecular-mechanics and cluster-models. Cations and solvent molecules in the 2nd coordination sphere affect the equatorial An←Oeq bonds more than the axial AnOax bonds. Common actinide contraction is found for calculated and experimental axial bond lengths of 92U to 94Pu, though no longer for 94Pu to 96Cm. The tendency of U to Pu forming actinyl(VI) species dwindles away toward Cm, which already features the preferred AnIII/LnIII oxidation state of the later actinides and all lanthanides. The well known change from d-type to typical U–Pu–Cm type and then to f-type behavior is labeled as the plutonium turn, a phenomenon that is caused by f-orbital energy-decrease and f-orbital localization with increase of both nuclear charge and oxidation state, and a non-linear variation of effective f-electron population across the actinide series. Both orbital and configuration mixing and occupation of antibonding 5f type orbitals increase, weakening the AnOax bonds and reducing the highest possible oxidation states of the later actinides.