Co-reporter:Michael McGovern, Kevin D. Dorfman and David C. Morse
Soft Matter 2016 vol. 12(Issue 29) pp:6214-6222
Publication Date(Web):24 Jun 2016
DOI:10.1039/C6SM00785F
We use Langevin dynamics simulations to study aggregation of semiflexible polymers driven by attractions between polymers and spherical particles. We consider a simple model with purely repulsive polymer/polymer and particle/particle interactions but attractive polymer/particle interactions. We find a rich “phase diagram” that contains several different types of globular and rod-like aggregates with either liquid-like or crystalline structure for the particle positions. Systems that exhibit rod-like aggregates with crystalline internal order exhibit a discontinuous rod-globule transition, while systems with liquid-like internal order exhibit a smooth crossover between isotropic and elongated aggregates with increasing chain stiffness. Polymers in elongated liquid-like aggregates often adopt helical configurations that wind around the axis of the aggregate.
Co-reporter:Pavani Medapuram, Jens Glaser, and David C. Morse
Macromolecules 2015 Volume 48(Issue 3) pp:819-839
Publication Date(Web):January 21, 2015
DOI:10.1021/ma5017264
We present a simulation study of how properties of symmetric diblock copolymers depend on the invariant degree of polymerization N̅, focusing on the vicinity of the order–disorder transition (ODT). Results from several coarse-grained simulation models are combined to cover a range of N̅ ≃ 200–8000 that includes most of the experimentally relevant range. Results are presented for the free energy per chain, the value of χeN at the ODT, the latent heat of transition, the layer spacing, the composition profile, and compression modulus in the ordered phase. Universality (i.e., model independence) is demonstrated by showing that equivalent results for all properties are obtained from corresponding thermodynamic states of different simulation models. Corresponding states of symmetric copolymers are states with equal values of the parameters χeN and N̅, where χe is an effective Flory–Huggins interaction parameter and N is a degree of polymerization. The underlying universality becomes apparent, however, only if data are analyzed using an adequate estimate of χe, which we obtain by fitting the structure factor in the disordered state to recent theoretical predictions. The results show that behavior near the ODT exhibits a different character at moderate and high values of N̅, with a crossover near N̅ ≃ 104. Within the range N̅ ≲ 104 studied here, the ordered and disordered phases near the ODT both contain strongly segregated domains of nearly pure A and B, in contrast to the assumption of weak segregation underlying the Fredrickson–Helfand (FH) theory. In this regime, the FH theory is inaccurate and substantially underestimates the value of χeN at the ODT. Results for the highest values of N̅ studied here agree reasonably well with FH predictions, suggesting that the theory may be accurate for N̅ ≳ 104. Self-consistent field theory (SCFT) grossly underestimates (χeN)ODT for modest N̅ because it cannot describe strong correlations in the disordered phase. SCFT is found, however, to yield accurate predictions for several properties of the ordered lamellar phase.
Co-reporter:Timothy M. Gillard, Pavani Medapuram, David C. Morse, and Frank S. Bates
Macromolecules 2015 Volume 48(Issue 8) pp:2801-2811
Publication Date(Web):April 14, 2015
DOI:10.1021/acs.macromol.5b00277
We present a detailed quantitative comparison of experimental results and theoretical predictions for the structure and thermodynamics of low molecular weight symmetric (fL ≈ 1/2) poly(1,4-isoprene-b-dl-lactide) (IL) diblock copolymers near the order–disorder transition (ODT). Small-angle neutron and X-ray scattering (SANS and SAXS) measurements obtained in the disordered phase with IL degree of polymerization N = 39 were fit to the renormalized one-loop (ROL) theory in order to estimate the effective interaction parameter χe(T). Calorimetric measurements of the latent heat of the ODT for the same copolymer compare well with that obtained from corresponding coarse-grained simulations, when the comparison is based on this estimate of χe(T). The corresponding estimate of (χeN)ODT at the experimental ODT of this polymer is much closer to the value obtained from simulations than to any theoretical prediction but differs from the simulation result by somewhat more than the bounds implied by experimental uncertainties. A larger discrepancy between simulation and experimental results for (χeN)ODT is obtained for longer chains, with N ≥ 50, when also calculated using χe(T, N = 39). We discuss possible reasons for this discrepancy, including the possibility of a significant end-group effect for these polymers. These results confirm the overwhelming importance of fluctuation effects in short diblock copolymers, and the usefulness of coarse-grained simulations as a starting point for quantitative modeling, but also indicate the need for attention to nonuniversal features of specific polymers that can also become more important with decreasing chain length.
Co-reporter:Kwanho Chang, Christopher W. Macosko, and David C. Morse
Macromolecules 2015 Volume 48(Issue 22) pp:8154-8168
Publication Date(Web):November 16, 2015
DOI:10.1021/acs.macromol.5b01268
The interfacial tension between polystyrene (PS) and polybutadiene (PB) homopolymers is measured in the presence of a nearly symmetric poly(styrene-b-butadiene) (SB) copolymer and compared to independent measurements of the critical micelle concentration (CMC) by transmission electron microscopy (TEM) and small-angle X-ray scattering (SAXS), and to self-consistent field theory (SCFT) predictions. Interfacial tension is measured with a spinning drop tensiometer (SDT) containing a drop of PB in a PS matrix, using samples in which varying concentrations of SB copolymer were initially added to either PS or PB. When SB is premixed with PS, the dependence of the interfacial tension γ upon copolymer concentration c is qualitatively similar to that expected in equilibrium, showing a decrease of γ with increasing c for c less than an apparent CMC and then saturating at higher concentrations. TEM and SAXS studies of a binary mixture of the same copolymer and PS homopolymer show, however, that the true CMC in PS is much lower than the apparent CMC inferred from these SDT experiments. We analyze the role of transport limitations in this experiment and propose that this discrepancy may be a result of a suppressed copolymer chemical potential near the interface due to continual diffusion of copolymer into the PB drop.
Co-reporter:Jens Glaser, Jian Qin, Pavani Medapuram, and David C. Morse
Macromolecules 2014 Volume 47(Issue 2) pp:851-869
Publication Date(Web):January 13, 2014
DOI:10.1021/ma401694u
We present a detailed comparison of simulations of disordered melts of symmetric AB diblock copolymers to predictions of the renormalized one-loop (ROL) theory. The behaviors of the structure factor S(q) and of single-chain correlations are studied over a range of chain lengths (N = 16, ..., 128) for two models: one with harshly repulsive pair interactions and another with very soft interactions. The ROL theory is shown to provide an excellent description of the dependence of S(q) on chain length and thermodynamic conditions for both models, even for very short chains, if we allow for the existence of a nonlinear dependence of the effective interaction parameter χe upon the strength of the AB repulsion. The decrease in peak wavenumber q* with increasing χe is shown to be unrelated to changes in single-chain correlations. Results for all quantities are consistent with the hypothesis that the ROL theory gives an exact description of the dominant corrections to RPA and random-walk predictions in the limit of infinite chain length N.
Co-reporter:Jens Glaser, Jian Qin, Pavani Medapuram, Marcus Müller and David C. Morse
Soft Matter 2012 vol. 8(Issue 44) pp:11310-11317
Publication Date(Web):13 Sep 2012
DOI:10.1039/C2SM26536B
Coarse-grained theories of dense polymer liquids such as block copolymer melts predict a universal dependence of equilibrium properties on a few dimensionless parameters. For symmetric diblock copolymer melts, such theories predict a universal dependence on only χN and , where χ is an effective interaction parameter, N is the degree of polymerization, and is a measure of overlap. We test whether simulation results for the structure factor S(q) obtained from several different simulation models are consistent with this two-parameter scaling hypothesis. We compare results from three models: (1) a lattice Monte Carlo model, the bond-fluctuation model, (2) a bead–spring model with harsh repulsive interactions, similar to that of Kremer and Grest, and (3) a bead–spring model with very soft repulsion between beads, and strongly overlapping beads. We compare results from pairs of simulations of different models that have been designed to have matched values of , over a range of values of χN and N, and devise methods to test the scaling hypothesis without relying on any prediction for how the phenomenological interaction parameter χ depends on more microscopic parameters. The results strongly support the scaling hypothesis, even for rather short chains, confirming that it is indeed possible to give an accurate universal description of simulation models that differ in many details.
.jpg)



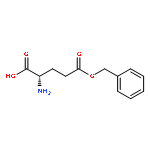
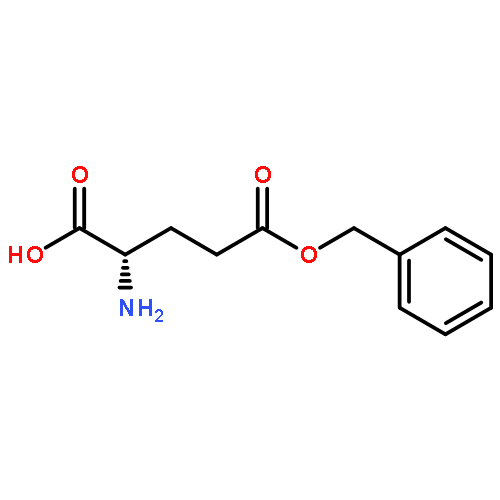
![Poly[imino[(2S)-1-oxo-2-[3-oxo-3-(phenylmethoxy)propyl]-1,2-ethanediyl
]]](http://img.cochemist.com/ccimg/25100/25038-53-3.png)
![Poly[imino[(2S)-1-oxo-2-[3-oxo-3-(phenylmethoxy)propyl]-1,2-ethanediyl
]]](http://img.cochemist.com/ccimg/25100/25038-53-3_b.png)