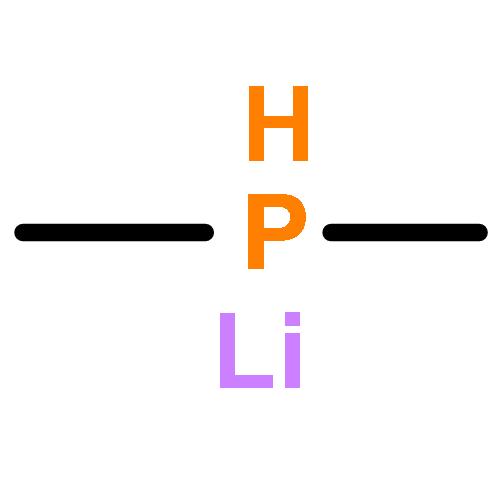
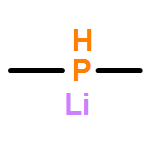Co-reporter:Dennis Wiedemann and Andreas Grohmann
Dalton Transactions 2014 vol. 43(Issue 6) pp:2406-2417
Publication Date(Web):26 Nov 2013
DOI:10.1039/C3DT53070A
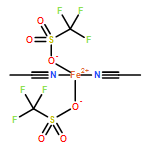
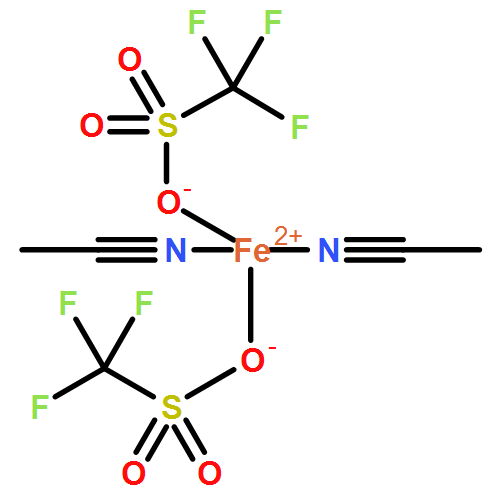
We have recently shown that the vacuum-deposited complex [FeIIL(NCS)2] (L: 1-{6-[1,1-di(pyridin-2-yl)ethyl]-pyridin-2-yl}-N,N-dimethylmethanamine) is capable of a thermally induced spin crossover (SCO) in direct contact with a graphite surface. The SCO significantly differs from the transition behaviour in the bulk phase (powder). In the present work, the assumption of virtually no intermolecular interaction in the powder is confirmed by comparison with the spin transition in acetone solution (T1/2 = 234[3] K, ΔT80 = 58[4] K), as monitored by temperature-dependent UV/Vis spectroscopy. The complex crystallises from chlorocarbons in the form of a number of pseudopolymorphs. Amongst these, the sufficiently stable solvate [FeIIL(NCS)2]·CHCl3 is investigated by variable-temperature single-crystal X-ray diffractometry. Its SCO behaviour (T1/2 = 240[3] K, ΔT80 = 35[4] K) correlates with features of molecular structure that are unambiguously identified by analysis of the tensor of thermal expansion. Following comprehensive comparison of spin-transition properties in different states of aggregation (also in relation to the newly synthesised high-spin iron(II) and iron(III) complexes [FeIICl2L] and [FeIIICl2L]PF6), a mode of adsorption on graphite surfaces is proposed, that complies with all previous findings.
Co-reporter:Anna Jozwiuk;E. Alper Ünal;Stefan Leopold;John P. Boyd;Marco Haryono;Nadine Kurowski;Francisco Velazquez Escobar;Peter Hildebrt;Jochen Lach;Frank W. Heinemann;Dennis Wiedemann;Elisabeth Irran
European Journal of Inorganic Chemistry 2012 Volume 2012( Issue 18) pp:3000-3013
Publication Date(Web):
DOI:10.1002/ejic.201101265
Abstract
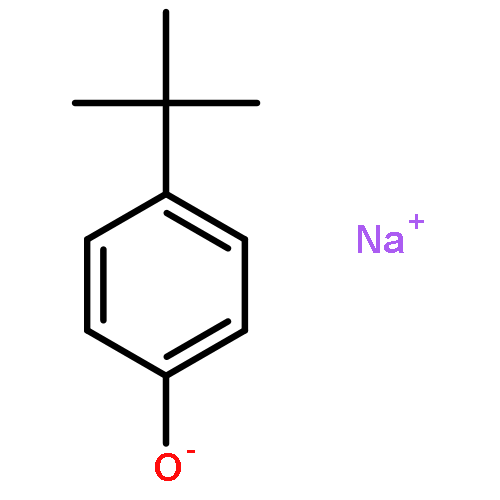
The results of studies focussed on copper complexes of a variety of ligands with an NN4 donor set are reported. The permethylated tetrapodal ligand 2 forms a complex with copper(I) which, upon reaction with dioxygen at –90 °C, yields a product having a bis(μ-oxido)dicopper(III) core (“O-type” product, 10), as inferred from UV/Vis and resonance-Raman spectroscopic data. The UV/Vis spectrum of 10 has two bands at 300 and 404 nm, with extinction coefficients of 9400 and 10400 L mol–1 cm–1, respectively. Resonance-Raman spectra display two 16O/18O-sensitive bands which, based on the isotopic shifts and the absolute frequencies, are attributed to the Cu–O stretching modes of the O-type product. Complex 10 shows tyrosinase-like activity, as its reaction with sodium p-tert-butylphenolate at –90 °C in THF yields p-tert-butylcatechol, in an ortho-hydroxylation reaction (yield: 30 %). Two new rigid tetrapodal pentadentate ligands (the “superpods” 3 and 4) can be synthesized by condensation of the primary polyamine 1 with paraformaldehyde. Their copper(II) complexes (5 and 6) have been spectroscopically characterized. As ascertained by X-ray crystallography, 5 has the CuII ion in a tetragonal-pyramidal environment, with almost uniform Cu–N bond lengths (basal bonds: 2.052 Å and 2.070 Å; apical bond: 2.077 Å). No significant Jahn–Teller distortion is observed here. In 6, the ligand acts as a multinucleating donor, which leads to the formation of a ladder-like cluster of [Cu(μ3-OH)] units containing a total of two ligands, six copper(II) ions, four hydroxido ligands and eight trifluoroacetate ions. Two of the trifluoroacetate ions are non-coordinating. Variable-temperature magnetic susceptibility data are reported for this hexanuclear copper(II) cluster. Copper(I) complexes of 1 and 3 have been characterized and allowed to react with molecular oxygen, which caused the decomposition of the complexes. The IR spectra of the oxygenation products have bands at 1652 and 1632 cm–1, respectively, which are absent in the spectra of 1 and 3, suggesting that amine functions have been oxidized to imines.
Co-reporter:Simon-Andreas Gentschow;Stephan W. Kohl;Walter Bauer;Markus Hummert
European Journal of Inorganic Chemistry 2011 Volume 2011( Issue 4) pp:556-566
Publication Date(Web):
DOI:10.1002/ejic.201001014
Abstract
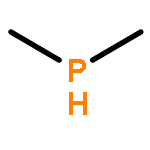
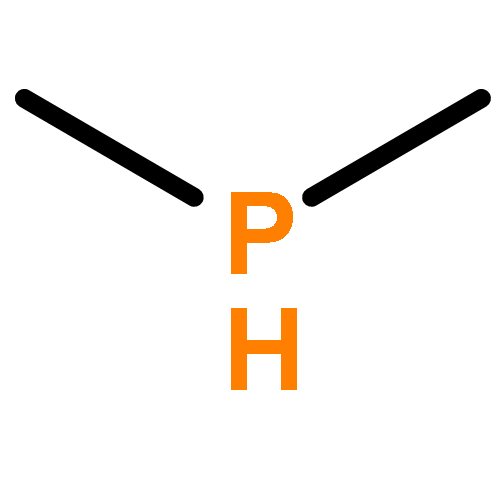
A mononuclear iron(II) complex is readily formed when combining equimolar amounts of iron(II) tetrafluoroborate hexahydrate, the pyridine-derived triphosphane C5H3N{2-[CMe(CH2PMe2)2]}{6-[CMe2(CH2PMe2)]} (2) and diethylphosphane (Et2PH) in methanol at room temperature. The chelate ligand is in fact pentadentate, as one of the methyl groups of the neopentyl-like sidearm engages in a C–H-bonded contact (agostic interaction) with the metal centre, in addition to the expected NP3 coordination. The remaining site of what is a distorted coordination octahedron is occupied by monodentate Et2PH. The autoclave reaction of this purple complex with CO (10 bar, ethanol solvent, 65 °C) yields a yellow microcrystalline precipitate, whose analysis reveals a mixture of two products, in an approximate ratio of 40:1. One of the products is the cis-dicarbonyl complex[Fe(2)(CO)2](BF4)2 (4), in which the chelate ligand acts as an NP3 donor, and the other is the monocarbonyl complex of the tetrapodal pentadentate NP4 ligand 1. The latter ligand is formed from 2 by incorporation of an additional PMe2 donor, in what, in effect, is a metal-mediated phosphenium group transfer. A mechanism is suggested for this reaction. Other species in the reaction mixture have been identified on the basis of mass spectra, and the full NMR spectroscopic assignment of complex 4 (1H, 31P, 13C) is reported.
Co-reporter:Marc Schmidt, Dennis Wiedemann, Andreas Grohmann
Inorganica Chimica Acta 2011 Volume 374(Issue 1) pp:514-520
Publication Date(Web):1 August 2011
DOI:10.1016/j.ica.2011.02.066
2-Methyl-2-(pyridin-2-yl)propane-1,3-diamine and formaldehyde are condensed to prepare the hexahydropyrimidine derivative, which is subsequently reacted with two equivalents of 2-vinylpyridine, to produce a novel, potentially pentadentate amine/imine ligand. Full NMR spectroscopic details are reported. The ligand, hexahydro-5-methyl-5-(pyridin-2-yl)-1,3-bis[2-(pyridin-2-yl)ethyl]pyrimidine, acts as a pentadentate in a series of first-row transition metal complexes (M = Ni2+, Fe2+, Zn2+, Cu2+) but is tridentate towards Mn2+, in the corresponding dibromido complex. Single-crystal X-ray structure analyses reveal the metal ions to be hexacoordinate in the case of M = Ni2+, Fe2+, with and additional aqua or halido (Br−, Cl−) ligand, or pentacoordinate (M = Zn2+, Cu2+, Mn2+). Ferric complexes were not obtained, neither from complexation experiments employing iron(III), nor from oxidations using the iron(II) complex, and hydrogen peroxide or iodosylbenzene. In the case of the latter reactions, mass spectrometric data indicate oxidation of the hexahydropyrimidine core, with concomitant decomplexation of the ligand.Graphical abstractA novel N5 ligand L imposes distorted coordination geometries on first-row transition metal ions, which can be hexacoordinate (in the presence of an additional monodentate ligand) or pentacoordinate. L readily forms complexes with ferrous but not ferric iron.Highlights► A novel triimino diamine ligand has been prepared. ► The ligand has a hexahydropyrimidine core. ► The coordination geometry of first-row transition metal complexes is distorted octahedral or square-pyramidal, as applicable. ► Iron(II) complexes are readily accessible. ► Iron(III) complexes proved inaccessible.
Co-reporter:Andreas Grohmann
Dalton Transactions 2010 vol. 39(Issue 6) pp:1432-1440
Publication Date(Web):22 Oct 2009
DOI:10.1039/B913436K
Tetrapodal pentadentate ligands occupy five coordination positions in a coordination octahedron, thereby providing the metal ion with a square-pyramidal “coordination cap”: In such complexes, all reactivity is focused on a single coordination site. The review highlights recent advances in the coordination chemistry of iron. With a variety of NN4 ligands, the concept is being used to model non-heme active sites in biomolecules. Tetraphosphane ligands (donor set: NP4) undergo, depending on the solvent, remarkably specific P–C bond activation reactions, which may be reversed under suitable conditions.
Co-reporter:Sophie K. Hain;Frank W. Heinemann;Klaus Gieb;Paul Müller;Gerald Hörner
European Journal of Inorganic Chemistry 2010 Volume 2010( Issue 2) pp:221-232
Publication Date(Web):
DOI:10.1002/ejic.200900588
Abstract
The temperature-dependent and photodynamic spin behaviour of three iron(II) complexes with different 2,6-dipyridyl-4-phenyltriazine ligands L1–L3 was investigated in the solid state and in solution (DMSO). The ligands differ in the substituent R in the 4-position of the phenyl ring (L1: R = H; L2: R = OCH3; L3: R = SAc), which allows the electronic properties of the ligands to be finetuned. The magnetic data for the complex [Fe(L3)2](BF4)2 in the solid state indicate an incomplete spin transition to the high-spin form upon warming from liquid helium temperature, reaching about 30 % at 400 K. There is circumstantial evidence for paramagnetic contributions compatible with spin transitions, namely, temperature-dependent NMR spectroscopic line shifts and line broadening in solution (DMSO). However, an efficient thermally induced spin crossover in solution is hindered by the substitution lability of the complexes, as has been detected and analyzed in an extended temperature-dependent UV/Vis spectroscopic study. Essentially unaffected by thermally induced substitution lability, the transient dynamics of the iron(II) complexes after nanosecond laser flash excitation of their metal-to-ligand charge-transfer bands provide good evidence for efficient photoinduced spin transitions in solution in all cases. The range of measured lifetimes of the high-spin quintet states is in accord with previously published data. Importantly, in our series of iron(II) complexes, the lifetimes of the high-spin state reflect the electron-donating character of the ligands.
Co-reporter:Sophie K. Hain;Frank W. Heinemann;Klaus Gieb;Paul Müller;Gerald Hörner
European Journal of Inorganic Chemistry 2010 Volume 2010( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/ejic.201090000
Abstract
The cover picture shows the crystal structure of an iron(II) complex with a tridentate triazine-based ligand, one of three complexes whose thermal and photonic spin-dynamics are discussed in the article by G. Hörner, A. Grohmann et al. on p. 221 ff. While there is a competition between spin crossover and ligand exchange upon thermal excitation in solution, the complexes switch cleanly between their stable low-spin and metastable high-spin forms upon photoexcitation. Artwork: Holger Neumann, Gentura.
Co-reporter:MohammadS. Alam Dr.;Michael Stocker;Klaus Gieb;Paul Müller Dr.;Marco Haryono Dr.;Katja Student Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 6) pp:1159-1163
Publication Date(Web):
DOI:10.1002/anie.200905062
Co-reporter:Marco Haryono;Frank W. Heinemann;Konstantin Petukhov;Klaus Gieb;Paul Müller
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 14) pp:2136-2143
Publication Date(Web):
DOI:10.1002/ejic.200900130
Abstract
The ligand 2,6-bis(1H-pyrazol-1-yl)-4-(thiocyanatomethyl)pyridine (L) has been prepared from the hydroxymethyl precursor by OH/Br exchange and subsequent thiocyanate substitution. Two polymorphs of the unsolvated iron(II) complex [Fe(L)2](BF4)2 crystallize at room temperature from the same solution (MeOH), and side by side. One (yellow) is high spin between 4 and 350 K, whereas the other (red-brown) shows spin crossover, with a thermal spin-transition centred at 272 K (width ca. 50 K). Both forms have been fully characterized by variable-temperature structure determination and magnetic susceptibility measurements. At a given temperature, the solid-state structures differ strongly with respect to the coordination geometry at iron(II), the orientation of the thiocyanatomethyl substituents, and cation-anion contacts. The implications of these structural differences for the observed spin behaviour are discussed. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Cai Shen, Marco Haryono, Andreas Grohmann, Manfred Buck, Tobias Weidner, Nirmalya Ballav and Michael Zharnikov
Langmuir 2008 Volume 24(Issue 22) pp:12883-12891
Publication Date(Web):October 24, 2008
DOI:10.1021/la8019974
Self-assembled monolayers (SAMs) of a bis(pyrazol-1-yl)pyridine-substituted thiol (bpp-SH) on Au (111)/mica were studied with scanning tunneling microscopy (STM), X-ray photoelectron spectroscopy (XPS), and near-edge X-ray absorption fine structure spectroscopy (NEXAFS). Using substrates precoated with perylene-3,4,9,10-tetracarboxylic acid dianhydride (PTCDA), preparation at elevated temperatures yields highly ordered layers whose structure is described by a rectangular (5 × √3) unit cell containing one molecule. The bis(pyrazol-1-yl)pyridine (bpp) units exhibit π-stacking along the ⟨112̅⟩ direction, and they are tilted significantly. We conclude the three imine nitrogen atoms in the bpp headgroup adopt a trans,trans arrangement.
Co-reporter:Jesús Pitarch López, Holger Kämpf, Matthias Grunert, Philipp Gütlich, Frank W. Heinemann, Raju Prakash and Andreas Grohmann
Chemical Communications 2006 (Issue 16) pp:1718-1720
Publication Date(Web):09 Mar 2006
DOI:10.1039/B517635B
Complexation of the tetrapodal pentadentate NN4 ligand 2,6-C5H3N[CMe(CH2NH2)2]2 (1) with iron(II) perchlorate hydrate in methanol, in the presence of N-methylimidazole, produces a diferrous complex with a single, unsupported μ-OH ligand between two {(1)FeII} coordination modules.
Co-reporter:Stephan W. Kohl, Frank W. Heinemann, Markus Hummert, Walter Bauer and Andreas Grohmann
Dalton Transactions 2006 (Issue 47) pp:5583-5592
Publication Date(Web):12 Oct 2006
DOI:10.1039/B610792C
The pyridine-derived tetrapodal tetraphosphane C5H3N[CMe(CH2PMe2)2]2 (1) is susceptible to selective protonolysis of a phosphorus–carbon bond in the presence of iron(II) salts. Water produces dimethylphosphinic acid, Me2POH, and protonates the anionic remainder of the tetraphosphane. The resulting iron(II) complexes 2 and 3 (tetrafluoroborate and perchlorate salts, respectively) contain the residual chelate ligand in which a methyl group, derived from the ligand skeleton, is in agostic interaction with the metal centre, and in which Me2POH, unavailable in the free state owing to rapid tautomerisation, is metal-coordinated and thus stabilised. Full NMR details are presented, including 31P simulations. The reactivity towards alcohols is similar (compounds 4, 5), and has been studied using deuterium labels (NMR). P–C bond cleavage may be suppressed only if all protic agents are rigorously excluded, as in the reaction of 1 with Fe(SO3CF3)2·2CH3CN in acetonitrile solution, which produces the complex [(1)Fe(NCMe)](SO3CF3)2 (6). In it, the ligand acts as an NP4 coordination cap but is severely distorted from square-pyramidal geometry. The reaction of 1 with anhydrous ferrous bromide, FeBr2, in methanol again produces a dimethylphosphinic acid ester ligand, but the complex (7) now contains ferric iron coordinated by a carbanionic residual chelate ligand, implicating H+ as the oxidising agent under these conditions. Full spectroscopic and X-ray structural details are presented for all compounds.
Co-reporter:Stephan W. Kohl;Frank W. Heinemann;Markus Hummert;Hardy Weißhoff
European Journal of Inorganic Chemistry 2006 Volume 2006(Issue 19) pp:
Publication Date(Web):10 AUG 2006
DOI:10.1002/ejic.200600530
The reaction of the 2,6-diethylpyridine-derived tetraphosphane ligand C5H3N[CMe(CH2PMe2)2]2 (1Me) with nickel(II) or cobalt(II) perchlorate or tetrafluoroborate in methanol produces complexes of approximate square-pyramidal geometry (NP4 coordination). The nickel complexes are diamagnetic, with NMR spectral features (31P) that reflect a tetrahedral distortion of diametrically opposite phosphorus donors; this is also found in the solid state. In terms of bond lengthsand angles, all complexes differ significantly from theiranalogues containing the phenyl-substituted ligand C5H3N[CMe(CH2PPh2)2]2. The ligand 1Me is readily and completely oxidised to the corresponding tetrakis(phosphane oxide) upon reaction with NO. Slow oxidation of the cobalt(II) tetrafluoroborate complex of 1Me with aerobic oxygen in acetonitrile produces a trinuclear cobalt complex containing two equivalents of partially oxidised ligand (C2-symmetrical; donor set: [PMe2]2[P(O)Me2]2). Four phosphane oxide oxygen atoms coordinate the central cobalt(II) ion in tetrahedral fashion, whereas the lateral cobalt(II) ions are in a square-pyramidal environment provided by two PMe2 donors and three acetonitrile ligands in each case. The reaction of the cobalt(II) perchlorate complex of 1Me with carbonmonoxide gives an octahedral, 19-valence-electron dicarbonyl complex in which one of the dimethylphosphanyl groups is uncoordinated. A structural relative of 1Me, C5H3N[CMe(CH2PMe2)2][CMe2(CH2PMe2)] (2), which contains one fewer dimethylphosphanyl donor, can be prepared in a straightforward manner from 2-ethyl-6-isopropylpyridine in a four-stage process. In a series of mononuclear nickel(II) complexes, 2 employs only its three phosphane donors which, together with a monodentate ligand (acetonitrile, acetamide, or bromide), provide a tetragonal coordination environment for the metal ion which is intermediate between square planar and tetrahedral. The acetamide complex is generated from the acetonitrile complex by slow hydrolysis of the coordinated ligand. Full spectroscopic details for the complexes, as well as X-ray structure analyses, are reported. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Stephan W. Kohl Dipl.-Chem.;Frank W. Heinemann Dr.;Markus Hummert Dr.;Walter Bauer Dr. Dr.
Chemistry - A European Journal 2006 Volume 12(Issue 16) pp:
Publication Date(Web):10 MAR 2006
DOI:10.1002/chem.200501577
Complex formation between FeX2⋅6 H2O (X=BF4 or ClO4) and the pyridine-derived tetrapodal tetraphosphane C5H3N[CMe(CH2PMe2)2]2 (1) in methanol proceeds with solvent-induced cleavage of one PMe2 group. Depending on the reaction temperature and the nature of the counterion, iron(II) is coordinated, in distorted square-pyramidal fashion, by the anionic remainder of the chelating ligand, C5H3N[CMe(CH2PMe2)2][CMe(CH2PMe2)(CH2−)] (NP3C− donor set: X=BF4, −50 °C: 2; X=ClO4, RT: 4) or its protonated form C5H3N[CMe(CH2PMe2)2][CMe(CH2PMe2)(CH3)], in which the methyl group is in agostic interaction with the metal centre (X=BF4, RT: 3; X=ClO4, +50 °C: 5). A monodentate phosphinite ligand Me2POMe, formed from the cleaved PMe2 group and methanol, completes the coordination octahedron in both cases. Working in CD3OD (X=BF4, RT) gives the deuterium-substituted analogue of 3, with ligands L(CH2D) (L=residual chelating ligand) and Me2POCD3. A mechanism for the observed phosphorus–carbon bond cleavage is suggested. Complex 2, when isolated at −50 °C, is stable in the solid state even at room temperature. The reaction of 2 in methanol with carbon monoxide (10.5 bar) at elevated temperature forms, in addition to as yet unidentified side products, the carbonyl complex [(1)Fe(CO)](BF4)2 (7), in which the previous PC bond cleavage has been reversed, reforming the original tetrapodal pentadentate NP4 ligand 1. All compounds have been fully characterised, including X-ray structure analyses in most cases.
Co-reporter:Christopher Zimmermann;Frank W. Heinemann
European Journal of Inorganic Chemistry 2005 Volume 2005(Issue 17) pp:
Publication Date(Web):2 AUG 2005
DOI:10.1002/ejic.200500281
The tetrapodal pentadentate NP4 ligand 2,6-C5H3N[CMe(CH2PPh2)2]2, which is readily prepared from the reaction of the corresponding tetrabromide with KPPh2, forms mononuclear complexes with nickel(II) and cobalt(II). In the case of nickel(II), the coordination mode adopted by the ligand is sensitive to the nature of the counterion: Use of NiCl2 gives a chloro complex as one of the products, which, as determined by X-ray crystallography, has a central ion coordinated by three phosphane and one chloro substituent in a distorted square planar fashion; by contrast, the reaction with Ni(BF4)2 gives a complex of approximate square-pyramidal geometry as the only product, in which all five atoms of the NP4 set are coordinated to the nickel ion, despite the steric bulk of the eight phenyl substituents. The same type of complex is obtained with Co(BF4)2 and Co(ClO4)2. Attempts to increase the coordination number of either complex beyond 5 have been unsuccessful. In particular, the cobalt(II) complex (17 valence electrons) is practically inert to reagents such as O2, NO, NO+, I2, Cl2 or H2. Full spectroscopic details for the complexes, as well as X-ray structure analyses, are reported. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2005)
Co-reporter:Andreas Grohmann
Dalton Transactions 2010 - vol. 39(Issue 6) pp:NaN1440-1440
Publication Date(Web):2009/10/22
DOI:10.1039/B913436K
Tetrapodal pentadentate ligands occupy five coordination positions in a coordination octahedron, thereby providing the metal ion with a square-pyramidal “coordination cap”: In such complexes, all reactivity is focused on a single coordination site. The review highlights recent advances in the coordination chemistry of iron. With a variety of NN4 ligands, the concept is being used to model non-heme active sites in biomolecules. Tetraphosphane ligands (donor set: NP4) undergo, depending on the solvent, remarkably specific P–C bond activation reactions, which may be reversed under suitable conditions.
Co-reporter:Dennis Wiedemann and Andreas Grohmann
Dalton Transactions 2014 - vol. 43(Issue 6) pp:NaN2417-2417
Publication Date(Web):2013/11/26
DOI:10.1039/C3DT53070A
We have recently shown that the vacuum-deposited complex [FeIIL(NCS)2] (L: 1-{6-[1,1-di(pyridin-2-yl)ethyl]-pyridin-2-yl}-N,N-dimethylmethanamine) is capable of a thermally induced spin crossover (SCO) in direct contact with a graphite surface. The SCO significantly differs from the transition behaviour in the bulk phase (powder). In the present work, the assumption of virtually no intermolecular interaction in the powder is confirmed by comparison with the spin transition in acetone solution (T1/2 = 234[3] K, ΔT80 = 58[4] K), as monitored by temperature-dependent UV/Vis spectroscopy. The complex crystallises from chlorocarbons in the form of a number of pseudopolymorphs. Amongst these, the sufficiently stable solvate [FeIIL(NCS)2]·CHCl3 is investigated by variable-temperature single-crystal X-ray diffractometry. Its SCO behaviour (T1/2 = 240[3] K, ΔT80 = 35[4] K) correlates with features of molecular structure that are unambiguously identified by analysis of the tensor of thermal expansion. Following comprehensive comparison of spin-transition properties in different states of aggregation (also in relation to the newly synthesised high-spin iron(II) and iron(III) complexes [FeIICl2L] and [FeIIICl2L]PF6), a mode of adsorption on graphite surfaces is proposed, that complies with all previous findings.



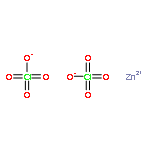

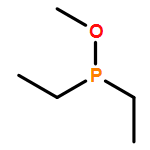
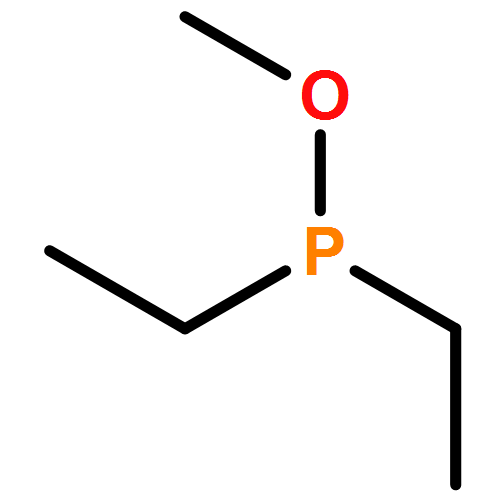
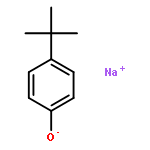



![PYRIDINE, 2,6-BIS[2-BROMO-1-(BROMOMETHYL)-1-METHYLETHYL]-](http://img.cochemist.com/ccimg/512800/512785-66-9.png)
![PYRIDINE, 2,6-BIS[2-BROMO-1-(BROMOMETHYL)-1-METHYLETHYL]-](http://img.cochemist.com/ccimg/512800/512785-66-9_b.png)