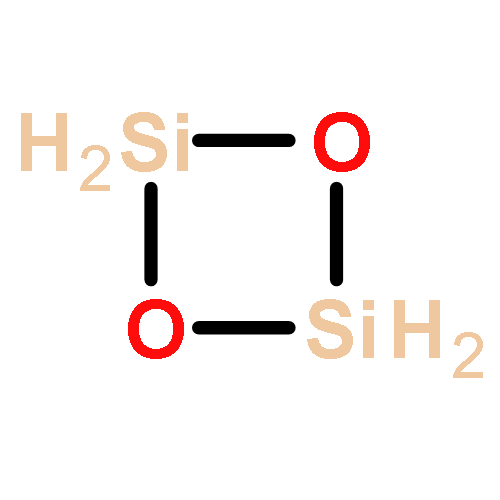
Co-reporter:Na Liu, Hui-Yan Zhao, Li-Jia Zheng, Sheng-Li Qin, Ying Liu
Chemical Physics Letters 2016 Volume 644() pp:219-224
Publication Date(Web):16 January 2016
DOI:10.1016/j.cplett.2015.11.046
Highlights•We calculated the geometric and electronic structures of metastable states of silicon oxide clusters using density functional theory.•The results show silicon-rich clusters and oxygen-rich clusters have different characteristics.•We calculated spectrums of the geometric structures to compare with observed spectra.•Possible structures which correspond to observed spectral lines are proposed.A systematic study of the geometric structures of steady states and metastable states of silicon oxide clusters has been performed using density functional theory. We find that silicon-rich and oxygen-rich clusters have different characteristics. Oxygen-rich clusters usually have oxygen atoms on the edges of the clusters, but separated from others by Si atoms. However, silicon-rich clusters tend to have rings nested within each other. The spectra for the structures have been calculated to compare with observed spectra. The predicted structures and spectroscopic properties are expected to be useful for the identification of silicon oxide species in the interstellar medium.Graphical abstract
Co-reporter:Jing Wang
Journal of Cluster Science 2016 Volume 27( Issue 3) pp:861-873
Publication Date(Web):2016 May
DOI:10.1007/s10876-015-0959-6
The present article summarizes progress in research on silicon clusters with encapsulated metal atoms, and specifically focuses on the recent identification of magnetic silicon fullerenes. Considering that C\(_{20}\) forms the smallest known fullerene, the Si\(_{20}\) cluster is of particular interest in this context. While the pure hollow Si\(_{20}\) cage is unstable due to the lack of \(sp^2\) hybridization, endohedral doping with a range of metal atoms has been considered to be an effective way to stabilize the cage structure. In order to seek out suitable embedded atoms for stabilizing Si\(_{20}\), a broad search has been made across elements with relatively large atomic radius. The rare earth elements have been found to be able to stabilize the Si\(_{20}\) cage in the neutral state by forming R@Si\(_{20}\) fullerene cages. Among these atoms, Eu@Si\(_{20}\) has been reported to yield a stable magnetic silicon fullerene. The central europium atom has a large magnetic moment of nearly 7.0 Bohr magnetons. In addition, based on a stable Eu\(_2\)Si\(_{30}\) tube, a magnetic silicon nanotube has been constructed and discussed.
These magnetic silicon fullerenes and nanotubes may have potential applications in the fields of spintronics and high-density magnetic storage.
Co-reporter:Hui-Yan Zhao, Jing Wang, Ling-Yan Ai, and Ying Liu
The Journal of Physical Chemistry A 2016 Volume 120(Issue 31) pp:6303-6308
Publication Date(Web):July 22, 2016
DOI:10.1021/acs.jpca.6b05258
A stable hollow copper silicide cage with Ih symmetry, Cu20Si12, constituted of a copper dodecahedron and a silicon icosahedron, was investigated using density functional theory. Molecular dynamics simulations show that Cu20Si12 retains its geometric topology up to an effective temperature of about 962 K. The molecule has a HOMO–LUMO gap of 1.099 eV, indicating its relatively high chemical stability. These frontier molecular orbitals show clear characteristics of hybridization between Si 3p and Cu 3d electrons. This proposed structure helps to extend the range of high-symmetry molecular polyhedral species. The hollow space within Cu20Si12 can be used to accommodate other atoms or molecules and emphasizes the benefit of studying endohedral fullerenes.
Co-reporter:Hong Zhang, Jing Wang, Zhi-Xue Tian and Ying Liu
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 21) pp:13821-13828
Publication Date(Web):12 Mar 2015
DOI:10.1039/C4CP06086E
A new class of Li–B sheets, along with the related nanotubes, with a Li2B5 primitive cell has been designed using first-principles density functional theory. The dynamical stability of the proposed structures was confirmed by calculation of the soft phonon modes, and the calculated electronic structures show that all are metallic. The application of both the sheets and nanotubes for hydrogen storage has been investigated and it has been found that both of them can adsorb two H2 molecules around each Li atom, with an average binding energy of 0.152–0.194 eV per H2, leading to a gravimetric density of 10.6 wt%.
Co-reporter:Hui-Yan Zhao ; Jing Wang ; Xiu-Jie Su ; Dong-Bo Zhang
The Journal of Physical Chemistry C 2014 Volume 118(Issue 47) pp:27502-27508
Publication Date(Web):November 4, 2014
DOI:10.1021/jp5080048
On the basis of the topologically distinct oxygen nets of crystalline ice phases, a series of carbon structures with sp3 bonding are constructed. Five new low-energy polymorphous phases of carbon, named “ice carbons”, are predicted by using the first-principles calculations. Their hardnesses are about 88.5–98.5% that of diamond, indicating that these new carbon phases are superhard materials. In particular, the new “iceIII-carbon” has the highest hardness 94.1 GPa that only 1.4 GPa smaller than that of diamond. Moreover, it also has slightly lower bulk modulus, which display similar properties with hP3, tI12, and tP12 superdense carbon allotropes.
Co-reporter:Dr. Jing Wang;Dr. Hui-Yan Zhao; Ying Liu
ChemPhysChem 2014 Volume 15( Issue 16) pp:3453-3459
Publication Date(Web):
DOI:10.1002/cphc.201402418
Abstract
Similar to carbon-based graphene, fullerenes and carbon nanotubes, boron atoms can form sheets, fullerenes, and nanotubes. Here we investigate several of these novel boron structures all based on the boron double ring within the framework of density functional theory. The boron sheet is found to be metallic and flat in its ground state. The spherical boron cage containing 180 atoms is also stable and has I symmetry. Stable nanotubes are obtained by rolling up the boron sheet, and all are metallic. The hydrogen storage capacity of boron nanostructures is also explored, and it is found that Li-decorated boron sheets and nanotubes are potential candidates for hydrogen storage. For Li-decorated boron sheets, each Li atom can adsorb a maximum of 4 H2 molecules with gd=7.892 wt %. The hydrogen gravimetric density increases to gd=12.309 wt % for the Li-decorated (0,6) boron nanotube.
Co-reporter:Dr. Jing Wang;Dr. Hui-Yan Zhao; Ying Liu
ChemPhysChem 2014 Volume 15( Issue 16) pp:
Publication Date(Web):
DOI:10.1002/cphc.201490077
Co-reporter:Hui-Yan Zhao, Jing Wang, Qing-Min Ma and Ying Liu
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 40) pp:17619-17625
Publication Date(Web):16 Sep 2013
DOI:10.1039/C3CP50946J
The Kelvin problem, how to partition three-dimensional space into cells of equal volume with minimal area, is a fascinating one. Aggregations of bubbles are naturally physical illustrations of the Kelvin problem. And the superconductor Na8Si46 as an inspiration leads to an amazing discovery of the Weaire–Phelan (WP) structure of foam – the optimal solution to the Kelvin problem to date. Here based on the structural similarity between sp3-bonded silicon allotropes and the solutions to the Kelvin problem, a series of new sp3-hybridization silicon allotropes, named “Kelvin Silicons”, are presented. Furthermore, the structural stability and electronic properties of these new silicon allotropes are investigated using density-functional theory (DFT) calculations. The results show that Kelvin Silicons are structurally stable semiconductors with indirect bandgaps in the range of 0.17–1.40 eV, and their bulk moduli are about 75.9–88.5% that of the diamond phase. The simulated X-ray diffraction spectra of the new silicon crystalline structures would provide more information for possible experimental observations and synthesis.
Co-reporter:Hui-Yan Zhao, Ke-Min Liu, Jing Wang, Hui-Yun Han, Ying Liu
Chemical Physics Letters 2013 Volume 555() pp:217-221
Publication Date(Web):3 January 2013
DOI:10.1016/j.cplett.2012.11.027
Abstract
How difficult is it for a guest atom to cut through C60 fullerene? Using an iron atom as a representative “intruder”, we explored the “quasistatic” processes of the atom going into and out from C60 along 3-fold or 5-fold axis and investigated the geometrical, electronic structures, and energy changes in the processes. Encaging an iron atom through 3-fold axis was more favored, with a lower energy barrier of about 4.20 eV. As it moved into C60 along 3-fold axis, the Fe atom must cross over two thresholds. The higher is located inside the C60 but not the outside.
Co-reporter:Jing Li ; Jing Wang ; Hui-Yan Zhao
The Journal of Physical Chemistry C 2013 Volume 117(Issue 20) pp:10764-10769
Publication Date(Web):April 29, 2013
DOI:10.1021/jp401090p
A europium-encapsulating silicon nanotube, Eu2@Si30, has been predicted by density functional theory. Electronic structure analysis shows that in Si–Si chemical bonds there exist sp2-like hybridizations induced by the europium atoms, and the hybridizations evidently enhance the stability of the silicon nanotube. Moreover, the nanotube of Eu2@Si30, with D5h symmetry, retains a high spin moment of 10 μB. On the basis of the Eu2Si30Tube, a stable magnetic silicon nanotube (SiNT) was obtained, and it is found to be metallic. Similar to the predictions and speculation of Daedalus, the new magnetic SiNT may have potential applications in the fields of spintronics and high-density magnetic storage.
Co-reporter:Hui-Yan Zhao, Li Wang, Jing Li, Ying Liu, You-Cheng Li, Sheng-Li Qin
Computational and Theoretical Chemistry 2012 Volume 994() pp:19-24
Publication Date(Web):15 August 2012
DOI:10.1016/j.comptc.2012.06.002
The candidate circumstellar molecule SiC4H was investigated using density functional theory at the B3LYP/6-311 + G(d, p) level. For the first three low-lying isomers, more accurate energies were obtained using single point energy calculations at the MP2/6-311 + G(2df, 2p) and CCSD (T)/6-311 + G(2df, 2p) levels. The results predict that the linear SiCCCCH isomer has the greatest stability. In addition, at the MP2/6-311 + G(2df, 2p) level of approximation, the rotational constant for the most stable isomer was predicted to be 1.40316 GHZ. The predicted structures and spectroscopic properties for the relevant isomers are expected to be useful for the identification of SiC4H and even larger SiCnH species in the laboratory and the interstellar medium.Graphical abstractHighlights► SiC4H was investigated as candidate circumstellar molecule. ► The linear SiCCCCH isomer has the greatest stability. ► IR spectrum of the most stable SiC4H isomer was predicted using the MP2 method.
Co-reporter:Zhi-Wei Zhao;Hui-Yan Zhao;Jing Wang;Qing-Min Ma
Journal of Cluster Science 2012 Volume 23( Issue 1) pp:133-145
Publication Date(Web):2012 March
DOI:10.1007/s10876-011-0436-9
The lowest-energy configurations, electronic structures and magnetic moments of small Lun (n = 2–20) clusters have been investigated within the framework of density functional theory. The results show that Lun (n = 4, 8, 13, and 18) clusters are more stable than their respective neighbors, and structural transformation reveals at n = 16. As the number of atoms increases, the magnetic moments increase in an alternating fashion until they reach a maximum of 4.00 μB for Lu8 clusters, followed by even–odd oscillation between 0.00 and 1.00 μB over the range n = 9–20.
Co-reporter:Jing Wang ; Shu-Shen Li ; Ying Liu ;Jingbo Li
The Journal of Physical Chemistry C 2012 Volume 116(Issue 39) pp:21039-21045
Publication Date(Web):September 5, 2012
DOI:10.1021/jp3048778
Using the first-principles band structure method, the electronic properties and optical properties of cupric iodide (CuI) quantum dots (QDs) are studied for the first time. A model is proposed to passivate the surface atoms of CuI QDs. In this model, pseudohydrogen atoms are used to passivate the dangling surface bonds, which remove the localized in-gap surface states. The size dependence of the QD gaps is obtained and is found to evolve as ΔEg = 1.60/d0.84 as the effective diameter d decreases. The energy of the calculated absorption peak is shifted higher with the decreasing d and the full width at half-maximum of the peak becomes larger as d increases, which are in good agreement with previous experiments. It is confirmed, although the local density approximation (LDA) calculations underestimate the band gap, that they give the trend of band gap shift as much as that obtained by the hybrid PBE0 for CuI QDs. These results provide understanding of the effects of the dimensionality of CuI nanocrystals, and it is expected that the method used in this work will be a practical approach to the study of other I–VII semiconductor nanocrystals.
Co-reporter:Qing-Min Ma, Zun Xie, Bao-Ru Wang, Ying Liu, You-Cheng Li
Solid State Communications 2011 Volume 151(Issue 10) pp:806-810
Publication Date(Web):May 2011
DOI:10.1016/j.ssc.2011.02.029
The geometries, electronic structures and magnetic moments of the FenCr (n=1–12n=1–12) clusters have been systematically investigated using all-electron density functional theory. For the lowest-energy structures of FenCr, the single Cr atom sits on the surface for all clusters up to n=10n=10. For n=11n=11 and n=12n=12 the Cr atom falls into the interior site. For FenCr (n=1–8,10–12n=1–8,10–12), the local moment of the Fe atoms is found to align antiferromagnetically with respect to that of the Cr atom, while for Fe9Cr, the local moments of the Fe atoms are ferromagnetic with respect to that of the Cr atom.Highlights► The growth behavior of FenCr(n=1–12)(n=1–12) clusters was studied using all-electron DFT. ► The single Cr atom sits on the surface for all clusters up to n=10n=10. ► For n=11n=11 and n=12n=12, the Cr atom falls into the interior site. ► The Cr atom has little effect on the original spin state for the Fen sub-clusters. ► The Fen sub-clusters and Cr atom are antiferromagnetically coupled except for Fe9Cr.
Co-reporter:Jing Wang, Ying Liu and You-Cheng Li
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 37) pp:11428-11431
Publication Date(Web):11 Aug 2010
DOI:10.1039/B923865D
A metal-encapsulating silicon fullerene, Eu@Si20, has been predicted by density functional theory to be by far the most stable fullerene-like silicon structure. The Eu@Si20 structure is a dodecahedron with D2h symmetry in which the europium atom occupies the center site. The calculated results show that the europium atom has a large magnetic moment of nearly 7.0 Bohr magnetons. In addition, it was found that a stable “pearl necklace” nanowire, constructed by concatenating a series of Eu@Si20 units, with the central europium atom, retains the high spin moment. The magnetic structure of the nanowire indicates potential applications in the fields of spintronics and high-density magnetic storage.
Co-reporter:Qing-Min Ma, Zun Xie, Ying Liu, You-Cheng Li
Solid State Communications 2010 Volume 150(31–32) pp:1439-1444
Publication Date(Web):August 2010
DOI:10.1016/j.ssc.2010.05.028
Co-reporter:Bao-Ru Wang, Qing-Min Ma, Ying Liu, You-Cheng Li
Solid State Communications 2009 Volume 149(5–6) pp:210-213
Publication Date(Web):February 2009
DOI:10.1016/j.ssc.2008.11.020
The effects of a manganese atom on the magnetic order and magnetic moments of FenMn (n=1–12n=1–12) clusters have been investigated using the all-electron density functional theory. The results reveal that the Fe–Mn couplings in the lowest-energy structures of FenMn (n=1–12n=1–12) clusters undergo a change from ferromagnetic ordering for the smallest (n=1,2n=1,2) clusters to ferrimagnetic ordering for the intermediate (n=3–6n=3–6) clusters. Starting from n=7n=7, however, ferromagnetic Fe–Mn couplings completely prevail. The low coordination number of the doped Mn atom results in it having a high spin moment in the Fe–Mn binary clusters, which exhibits a marked magnetic moment surface enhancement effect.
Co-reporter:Jing Wang Dr. ;You-Cheng Li
ChemPhysChem 2009 Volume 10( Issue 17) pp:3119-3121
Publication Date(Web):
DOI:10.1002/cphc.200900632
Abstract
The configurations, stability and electronic structures of a new class of boron sheet and related boron nanotubes are predicted within the framework of density functional theory. This boron sheet is sparser than those of recent proposals. Our theoretic results show that the stable boron sheet remains flat and is metallic. There are bands similar to the π-bands in graphite near the Fermi level. Stable nanotubes with various diameters and chiral vectors can be rolled from the sheet. Within our study, only the thin (8, 0) nanotube with a band gap of 0.44 eV is semiconducting, while all the other thicker boron nanotubes are metallic, independent of their chirality. It indicates the possibility, in the design of nanodevices, to control the electronic transport properties of the boron nanotube through the diameter.
Co-reporter:Jing Wang Dr. ;You-Cheng Li
ChemPhysChem 2009 Volume 10( Issue 17) pp:
Publication Date(Web):
DOI:10.1002/cphc.200990071
Co-reporter:Bao-Ru Wang, Jing Wang, Qing-Ming Ma, Ying Liu
Solid State Communications 2008 Volume 147(1–2) pp:53-56
Publication Date(Web):July 2008
DOI:10.1016/j.ssc.2008.04.020
The structures and magnetic ordering of MnnFe (n=1–12) clusters have been investigated using all-electron density functional theory. The results indicate that the MnnFe clusters undergo a change in magnetic behavior from ferromagnetic ordering for the smallest size to ferrimagnetic ordering for intermediate sizes and beyond. Ferromagnetic ordering is clearly favored for n=1n=1, but ferromagnetic and ferrimagnetic states are nearly degenerate for n=2n=2, 3, and 4. A radical change occurs at n=5n=5 where the ferrimagnetic states completely prevail. The transition range of magnetic ordering from ferromagnetic to ferrimagnetic for Mnn clusters occurs early with the doping of one Fe atom.
Co-reporter:Qing-Min Ma, Zun Xie, Jing Wang, Ying Liu, You-Cheng Li
Solid State Communications 2007 Volume 142(1–2) pp:114-119
Publication Date(Web):April 2007
DOI:10.1016/j.ssc.2006.12.023
The structures, binding energies, and magnetic moments of FeN(N=2–13,15,19) clusters have been obtained by all-electron density functional theory. The Jahn–Teller effect plays an important role in determining the ground state of certain geometric structures. For Fe3 and Fe4, new ground states are found. The results indicate that the magnetic moment per atom shows only small variations with cluster size and remains in the vicinity of 3.0μB/atom over this size range. With increasing atom number the mean binding energy monotonically decreases and the second derivative of the binding energy indicates that N=6N=6 and N=10N=10 are magic numbers for neutral FeN clusters.
Co-reporter:Qing-Min Ma, Zun Xie, Bao-Ru Wang, Ying Liu, You-Cheng Li
Solid State Communications (May 2011) Volume 151(Issue 10) pp:806-810
Publication Date(Web):1 May 2011
DOI:10.1016/j.ssc.2011.02.029
The geometries, electronic structures and magnetic moments of the FenCr (n=1–12) clusters have been systematically investigated using all-electron density functional theory. For the lowest-energy structures of FenCr, the single Cr atom sits on the surface for all clusters up to n=10. For n=11 and n=12 the Cr atom falls into the interior site. For FenCr (n=1–8,10–12), the local moment of the Fe atoms is found to align antiferromagnetically with respect to that of the Cr atom, while for Fe9Cr, the local moments of the Fe atoms are ferromagnetic with respect to that of the Cr atom.Highlights► The growth behavior of FenCr(n=1–12) clusters was studied using all-electron DFT. ► The single Cr atom sits on the surface for all clusters up to n=10. ► For n=11 and n=12, the Cr atom falls into the interior site. ► The Cr atom has little effect on the original spin state for the Fen sub-clusters. ► The Fen sub-clusters and Cr atom are antiferromagnetically coupled except for Fe9Cr.
Co-reporter:Ling-Yan Ai, Hui-Yan Zhao, Jing Wang, Ying Liu
Solid State Communications (March 2017) Volume 253() pp:1-5
Publication Date(Web):March 2017
DOI:10.1016/j.ssc.2017.01.023
Co-reporter:Bao-Ru Wang, Qing-Min Ma, Ying Liu, You-Cheng Li
Solid State Communications (February 2009) Volume 149(5–6) pp:210-213
Publication Date(Web):1 February 2009
DOI:10.1016/j.ssc.2008.11.020
The effects of a manganese atom on the magnetic order and magnetic moments of FenMn (n=1–12) clusters have been investigated using the all-electron density functional theory. The results reveal that the Fe–Mn couplings in the lowest-energy structures of FenMn (n=1–12) clusters undergo a change from ferromagnetic ordering for the smallest (n=1,2) clusters to ferrimagnetic ordering for the intermediate (n=3–6) clusters. Starting from n=7, however, ferromagnetic Fe–Mn couplings completely prevail. The low coordination number of the doped Mn atom results in it having a high spin moment in the Fe–Mn binary clusters, which exhibits a marked magnetic moment surface enhancement effect.
Co-reporter:Qing-Min Ma, Zun Xie, Ying Liu, You-Cheng Li
Solid State Communications (August 2010) Volume 150(31–32) pp:1439-1444
Publication Date(Web):1 August 2010
DOI:10.1016/j.ssc.2010.05.028
The geometries, binding energies and magnetic moments of CrMCN (M=1,2,N=1–8) clusters have been calculated using all-electron density functional theory. The CrCN (N=1,3,5–7) clusters prefer linear structures with the Cr atom at one end, while those with N=2,4,8 prefer cyclic planar structures. The lowest-energy structures of Cr2CN (N=1–8) are linear geometries with the two Cr atoms at the two ends. For all the clusters, Mulliken population analysis shows charge transfer from the Cr atom(s) to the C atoms with the magnetic moment lying mostly on the Cr atom(s).
Co-reporter:Hong Zhang, Jing Wang, Zhi-Xue Tian and Ying Liu
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 21) pp:NaN13828-13828
Publication Date(Web):2015/03/12
DOI:10.1039/C4CP06086E
A new class of Li–B sheets, along with the related nanotubes, with a Li2B5 primitive cell has been designed using first-principles density functional theory. The dynamical stability of the proposed structures was confirmed by calculation of the soft phonon modes, and the calculated electronic structures show that all are metallic. The application of both the sheets and nanotubes for hydrogen storage has been investigated and it has been found that both of them can adsorb two H2 molecules around each Li atom, with an average binding energy of 0.152–0.194 eV per H2, leading to a gravimetric density of 10.6 wt%.
Co-reporter:Jing Wang, Ying Liu and You-Cheng Li
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 37) pp:NaN11431-11431
Publication Date(Web):2010/08/11
DOI:10.1039/B923865D
A metal-encapsulating silicon fullerene, Eu@Si20, has been predicted by density functional theory to be by far the most stable fullerene-like silicon structure. The Eu@Si20 structure is a dodecahedron with D2h symmetry in which the europium atom occupies the center site. The calculated results show that the europium atom has a large magnetic moment of nearly 7.0 Bohr magnetons. In addition, it was found that a stable “pearl necklace” nanowire, constructed by concatenating a series of Eu@Si20 units, with the central europium atom, retains the high spin moment. The magnetic structure of the nanowire indicates potential applications in the fields of spintronics and high-density magnetic storage.
Co-reporter:Hui-Yan Zhao, Jing Wang, Qing-Min Ma and Ying Liu
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 40) pp:NaN17625-17625
Publication Date(Web):2013/09/16
DOI:10.1039/C3CP50946J
The Kelvin problem, how to partition three-dimensional space into cells of equal volume with minimal area, is a fascinating one. Aggregations of bubbles are naturally physical illustrations of the Kelvin problem. And the superconductor Na8Si46 as an inspiration leads to an amazing discovery of the Weaire–Phelan (WP) structure of foam – the optimal solution to the Kelvin problem to date. Here based on the structural similarity between sp3-bonded silicon allotropes and the solutions to the Kelvin problem, a series of new sp3-hybridization silicon allotropes, named “Kelvin Silicons”, are presented. Furthermore, the structural stability and electronic properties of these new silicon allotropes are investigated using density-functional theory (DFT) calculations. The results show that Kelvin Silicons are structurally stable semiconductors with indirect bandgaps in the range of 0.17–1.40 eV, and their bulk moduli are about 75.9–88.5% that of the diamond phase. The simulated X-ray diffraction spectra of the new silicon crystalline structures would provide more information for possible experimental observations and synthesis.