Co-reporter:Ruoning Wang, Ziqiang Zhao, Yue Han, Shihao Hu, Yaw Opoku-Damoah, Jianping Zhou, Lifang Yin, and Yang Ding
Molecular Pharmaceutics September 5, 2017 Volume 14(Issue 9) pp:2999-2999
Publication Date(Web):July 28, 2017
DOI:10.1021/acs.molpharmaceut.7b00192
The effective combination of drugs promoting antiangiogenesis and apoptosis effects has proven to be a promising collaborative tumor antidote; and the codelivery of small interfering RNA (siRNA) and chemotherapy agents within one efficient vehicle has gained more attention over single regimen administration. Herein, vascular endothelial growth factor specific siRNA (siVEGF) and paclitaxel (PTX) were introduced as therapeutic companions and coencapsulated into naturally mimic high-density lipoproteins (rHDL/siVEGF-PTX), so that various mechanisms of treatment can occur simultaneously. The terminal nanoparticles share capacity of specific-targeting to tumor cells overexpressed scavenger receptor class B type I (SR-BI) and deliver siVEGF and PTX into cytoplasm by a nonendocytosis mechanism. By exchanging HDL core lipids with hydrophobic therapeutics, rHDL/siVEGF-PTX possessed particle size of ∼160 nm, surface potential of ∼−20 mV, and desirable long-term storage stability. In vitro results confirmed that the parallel activity of siVEGF and PTX displayed enhanced anticancer efficacy. The half-maximal inhibitory concentration (IC50) of rHDL/siVEGF-PTX toward human breast cancer MCF-7 cell is 0.26 μg/mL (PTX concentration), which presents a 14.96-fold increase in cytotoxicity by taking Taxol as comparison. Moreover, in vivo results further demonstrated that rHDL/siVEGF-PTX performed enhanced tumor growth inhibition via natural targeting pathway, accompanied by remarkable inhibition of neovascularization in situ caused by siVEGF silencing in down-regulation of VEGF proteins. On the premise of effective drug codelivery, rHDL/siVEGF-PTX demonstrated high tumor targeting for collaborative antitumor efficacy without side effects after systemic administration, and this bioinspired strategy could open an avenue for exploration of combined anticancer therapy.Keywords: codelivery of siRNA and paclitaxel; collaborative antitumor efficacy; direct cytosolic delivery; reconstituted high density lipoproteins; vascular endothelial growth factor;
Co-reporter:Yang Ding, Yue Han, Ruoning Wang, Yazhe Wang, Cheng Chi, Ziqiang Zhao, Huaqing Zhang, Wei Wang, Lifang Yin, and Jianping Zhou
ACS Applied Materials & Interfaces September 13, 2017 Volume 9(Issue 36) pp:30488-30488
Publication Date(Web):August 22, 2017
DOI:10.1021/acsami.7b10047
High-density lipoprotein (HDL) is an outstanding biocompatible nanovector for tumor-targeted delivery of multimodel drugs in cancer therapy. However, this seemingly promising delivery platform demonstrates an adverse accumulation in liver and adrenal due to the primary expression of natural target scavenger receptor class B type I (SR-BI), which overexpressed in malignant cells as well. Therefore, we endowed native HDLs with rerouting capacity, that is, enabling HDLs to get away from natural receptors (SR-BI) to selectively alternate tumor-rich receptors. The αvβ3-integrin specific cyclic-RGDyk peptide was conjugated with HDL-protein component apolipoprotein A-I (apoA-I), demonstrating high substitution degree of 26.2%. Afterward, RGD-modified apoA-I was introduced to fabricate cholesterol siRNA-loaded HDL nanoparticles (RGD-HDL/Ch-siRNA) for specific affinity with tumor angiogenesis and αvβ3 integrin on tumor surface. After preparation, RGD-HDL/Ch-siRNA shared desirable particle size, efficient siRNA protection during blood circulation, and favorable proton sponge effect. αvβ3 integrin-associated superior rerouting capacity, endocytosis pathway, and rapid endolysosome escape were confirmed both in vitro and in vivo. For targeted gene silencing therapy, Pokemon-specific siRNA (siPokemon) was introduced as RNA interference candidate; the enhanced antitumor efficacy and decreased Pokemon expression level were commendably confirmed by tumor growth inhibition, survival period extension, and western blot analysis. Collectively, cyclic-RGDyk modification endows native HDLs with rerouting capacity to specific αvβ3 integrin receptor, which provides a promising strategy to extend malignancy targeting potential of native HDL to a broader purview.Keywords: cyclic-RGDyk peptide; enhanced gene silencing therapy; high-density lipoproteins; Pokemon-specific siRNA; rerouting capacity;
Co-reporter:Huaqing Zhang;Weiguang Chen;Ziqiang Zhao
Journal of Pharmaceutical Innovation 2017 Volume 12( Issue 3) pp:271-280
Publication Date(Web):02 May 2017
DOI:10.1007/s12247-017-9287-8
We describe here a novel lyophilized nanosuspension technology in order to improve the dissolution rate and oral bioavailability of the insoluble drug P2X7 receptor antagonist (PRA), which is an effective antagonist to P2X7 receptor for non-steroidal anti-inflammatory.PRA-lyophilized nanosuspension (PRA-LNS) was fabricated by anti-solvent precipitation in combination with high pressure homogenization, and then lyophilized for prolonged storage. After preparations, various characterization experiments were performed including particle size, zeta potential, surface morphology, X-ray powder diffraction (XRPD), differential scanning calorimetry (DSC), in vitro dissolution study, and in vivo pharmacokinetic study.The re-dissolved particle size of PRA-LNS was about 180~250 nm with uniform distribution, confirmed by TEM image. The drug PRA in nanosuspensions possessed crystalline form evaluated via XRPD and DSC analysis. The solubility of PRA-LNS in water was 1.52 times larger than PRA raw drug; in vitro dissolution tests showed that PRA-LNS could dissolve completely within 5 min, which is a significant improvement compared to the raw drug. The relative bioavailability of PRA-LNS is 290.70% compared to the raw drug and 177.94% compared to the physical mixture.PRA-LNS could easily re-disperse in water with increased solubility, enhanced oral bioavailability, and controllable production process.
Co-reporter:Jiali He;Yue Han;Gujun Xu;Lifang Yin;M. Ngandeu Neubi;Yang Ding
RSC Advances (2011-Present) 2017 vol. 7(Issue 22) pp:13053-13064
Publication Date(Web):2017/02/24
DOI:10.1039/C6RA28676C
Celecoxib (CLX) is a selective cyclooxygenase-2 (COX-2) inhibitor; however, the application of CLX is compromised due to its poor aqueous solubility and low oral bioavailability. This study aims to develop a nanotechnology to overcome these issues. Celecoxib nanosuspensions (CLX-NS) were formulated using D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) as stabilizer via high pressure homogenization (HPH) and then freeze-dried to solid powder. Transmission electron microscopy (TEM), differential scanning calorimetry (DSC), X-ray powder diffraction (XRD) and Fourier transform infrared spectroscopy (FT-IR) were employed to characterize the physicochemical and pharmaceutical properties of CLX-NS lyophilization. In vitro dissolution, an in situ single-pass intestinal perfusion study, and in vivo pharmacokinetic studies were performed for further investigation. Particles of the nanosuspensions were short-rod shaped (∼388 nm) and remained in the same crystalline state as CLX coarse powder. In the test of in vitro dissolution, CLX-NS displayed a higher dissolution amount (90.8% during 60 minutes) compared to groups of CLX coarse powder (47.9%) and physical mixture (52.9%). Moreover, in situ single-pass intestinal perfusion study indicated that CLX-NS could be well absorbed in the whole intestine with the main absorption site being the duodenum. Significant improvements in Cmax and AUC0–t of CLX-NS were obtained in pharmacokinetic analyses with a 245.8% increase in relative bioavailability compared to CLX coarse powder. This study showed that nanosuspension preparations could be a promising strategy in influencing CLX absorption in gastrointestinal tract, and improving dissolution and oral bioavailability of poorly water-soluble drugs.
Co-reporter:Ruoning Wang, Xiaochen Gu, Jianping Zhou, Lingjia Shen, Lifang Yin, Peiying Hua, Yang Ding
Journal of Controlled Release 2016 Volume 235() pp:134-146
Publication Date(Web):10 August 2016
DOI:10.1016/j.jconrel.2016.05.055
In this study, a simple and green approach ‘bioinspired disassembly-reassembly strategy’ was employed to reconstitute lipoprotein nanoparticles (RLNs) using whole-components of endogenous ones (contained dehydrated human lipids and native apolipoproteins). These RLNs were engineered to mimic the configuration and properties of natural lipoproteins for efficient drug delivery. In testing therapeutic targeting to microtubules, paclitaxel (PTX) was reassembled into RLNs to achieve improved targeted anti-carcinoma treatment and minimize adverse effects, demonstrating ultimately more applicable than HDL-like particles which are based on exogenous lipid sources. We have characterized that apolipoprotein-decoration of PTX-loaded RLNs (RLNs-PTX) led to favoring uniformly dispersed distribution, increasing PTX-encapsulation with a sustained-release pattern, while enhancing biostability during blood circulation. The innate biological RLNs induced efficient intracellular trafficking of cargos in situ via multi-targeting mechanisms, including scavenger receptor class B type I (SR-BI)-mediated direct transmembrane delivery, as well as other lipoprotein-receptors associated endocytic pathways. The resulting anticancer treatment from RLNs-PTX was demonstrated a half-maximal inhibitory concentration of 0.20 μg/mL, cell apoptosis of 18.04% 24 h post-incubation mainly arresting G2/M cell cycle in vitro, and tumor weight inhibition of 70.51% in vivo. Collectively, green-step assembly-based RLNs provided an efficient strategy for mediating tumor-targeted accumulation of PTX and enhanced anticancer efficacy.A “bioinspired disassembly-reassembly strategy” was applied for RLNs-PTX preparation via an emulsion-evaporation method by using isolated human lipids and original proportions of endogenous apolipoproteins, thereby achieving efficient drug accumulation in cancer cells via multiple pathways, such as SR-BI-mediated direct translocation as well as LDLR- or LRP-induced endocytosis.
Co-reporter:Shu Wang;Weiqin Wang;Huixia Lv
Journal of Pharmaceutical Innovation 2016 Volume 11( Issue 2) pp:134-142
Publication Date(Web):2016 June
DOI:10.1007/s12247-016-9246-9
Deoxypodophyllotoxin (DPT) is a new potential anti-tumor drug with nearly no water solubility and low permeability. Hence, we prepared a poly (ethylene glycol)-distearoylphosphatidylethanolamine (PEG-DSPE) modified long-circulation liposomes for enhanced solubility and anti-tumor capacity of DPT with clinical expectation.DPT-loaded long-circulation liposomes (DPT-LCLPs) were prepared by thin-film dispersion method, and characteristics including particle size, zeta potential and entrapment efficiency of re-dissolution after lyophilization were 110.5 ± 4.5 nm, −15.06 ± 1.14 mV, and 92.45 ± 5.21 %, respectively. TEM images showed that DPT-LCLPs appeared as spherical or ellipsoidal in shape with multilayer membrane. Moreover, sensitive liquid chromatography method was developed for quantification of DPT concentration in rat plasma with diazepam as internal standard (IS), and the results revealed that two-compartment intravenous model analysis was better with respect to data fitted by Kinetica 4.4 program. The terminal phase half-life (T1/2β) of DPT-LCLPs group was estimated to be approximately 155 min; AUC0−τ was more than three times higher than that of control group, demonstrating a prolonged circulation time due to PEG modification. In vivo investigation on Heps tumor-xenografted mice, DPT-LCLPs group indicated higher anti-tumor efficacy with dose-dependence, comparing to Etoposide and control group.PEG-modified liposomes highly improved the solubility and blood circulation of DPT with simple preparation for potentially enhanced anti-cancer therapy.
Co-reporter:Fanfei Meng, Sajid Asghar, Yurui Xu, Jianping Wang, Xin Jin, Zhilin Wang, Jing Wang, Qineng Ping, Jianping Zhou, Yanyu Xiao
International Journal of Pharmaceutics (15 June 2016) Volume 506(Issues 1–2) pp:46-56
Publication Date(Web):15 June 2016
DOI:10.1016/j.ijpharm.2016.04.033
Many nanoparticle matrixes have been demonstrated to be efficient in brain targeting, but there are still certain limitations for them. To overcome the shortcomings of the existing nanoparticulate systems for brain-targeted delivery, a lipoprotein resembling protein-free nanostructured lipid carrier (PS80-NLC) loaded with curcumin was constructed and assessed for in vitro and in vivo performance. Firstly, single factor at a time approach was employed to investigate the effects of various formulation factors. Mean particle sizes of ≤100 nm, high entrapment efficiency (EE, about 95%) and drug loading (DL, >3%) were obtained for the optimized formulations. In vitro release studies in the presence of plasma indicated stability of the formulation under physiological condition. Compared with NLC, PS80-NLC showed noticeably higher affinity for bEnd.3 cells (1.56 folds greater than NLC) but with lower uptake in macrophages. The brain coronal sections showed strong and widely distributed fluorescence intensity of PS80-NLC than that of NLC in the cortex. Ex vivo imaging studies further confirmed that PS80-NLC could effectively permeate BBB and preferentially accumulate in the brain (2.38 times greater than NLC). The considerable in vitro and in vivo performance of the safe and biocompatible PS80–NLC makes it a suitable option for further investigations in brain targeted drug delivery.Download high-res image (185KB)Download full-size image




![1H-PYRIDAZINO[1,2-A]INDAZOL-1-ONE, 6,7,8,9-TETRAHYDRO-3-METHYL-](http://img.cochemist.com/ccimg/701300/701203-91-0.png)
![1H-PYRIDAZINO[1,2-A]INDAZOL-1-ONE, 6,7,8,9-TETRAHYDRO-3-METHYL-](http://img.cochemist.com/ccimg/701300/701203-91-0_b.png)










![6H-Furo[3,4-g]-1-benzopyran-6-one,2,8-dihydro-5-hydroxy-2,2-dimethyl-](http://img.cochemist.com/ccimg/165500/165467-63-0.png)
![6H-Furo[3,4-g]-1-benzopyran-6-one,2,8-dihydro-5-hydroxy-2,2-dimethyl-](http://img.cochemist.com/ccimg/165500/165467-63-0_b.png)






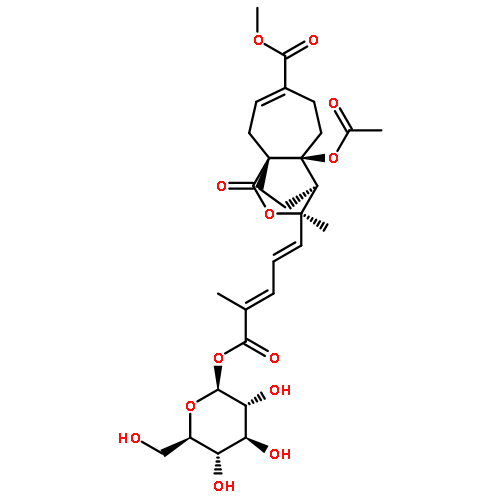


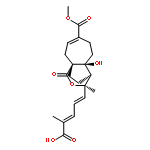
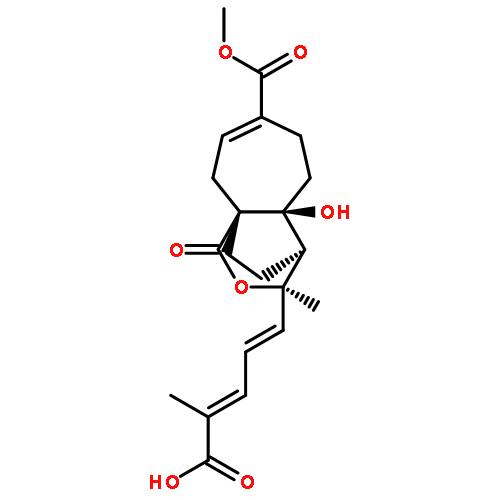
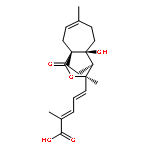




![Benzeneacetonitrile, a-[3-[[2-(3,4-dimethoxyphenyl)ethyl]amino]propyl]-3,4-dimethoxy-a-(1-methylethyl)-](http://img.cochemist.com/ccimg/67100/67018-85-3.png)
![Benzeneacetonitrile, a-[3-[[2-(3,4-dimethoxyphenyl)ethyl]amino]propyl]-3,4-dimethoxy-a-(1-methylethyl)-](http://img.cochemist.com/ccimg/67100/67018-85-3_b.png)
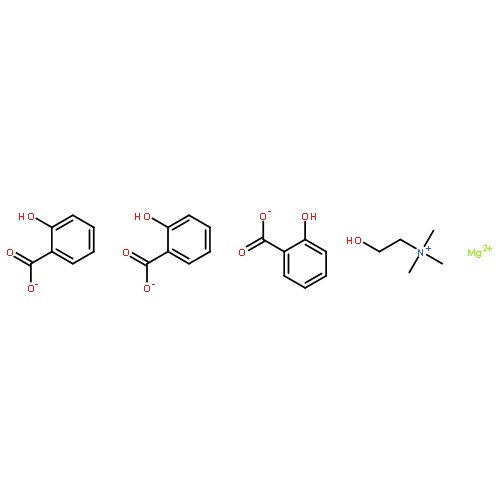
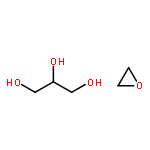

![4H-1-Benzopyran-4-one, 2-(3,4-dihydroxyphenyl)-3-[(O-β-D-glucopyranosyl-(1→2)-O-β-D-glucopyranosyl-(1→2)-β-D-glucopyranosyl)oxy]-5,7-](/data/chemimg/1817600/38681-85-5.png)
![4H-1-Benzopyran-4-one, 2-(3,4-dihydroxyphenyl)-3-[(O-β-D-glucopyranosyl-(1→2)-O-β-D-glucopyranosyl-(1→2)-β-D-glucopyranosyl)oxy]-5,7-](/data/chemimg/1817600/38681-85-5_b.png)









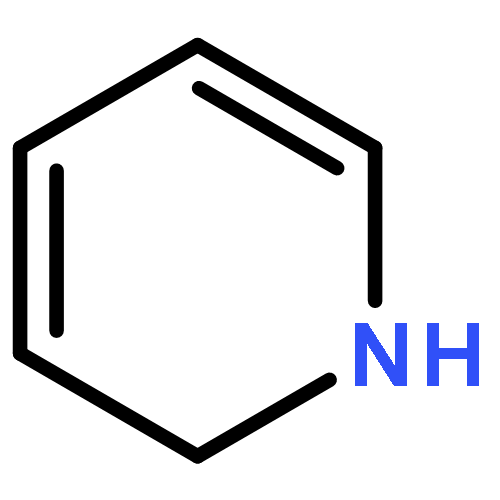


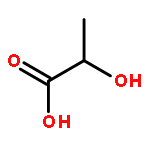
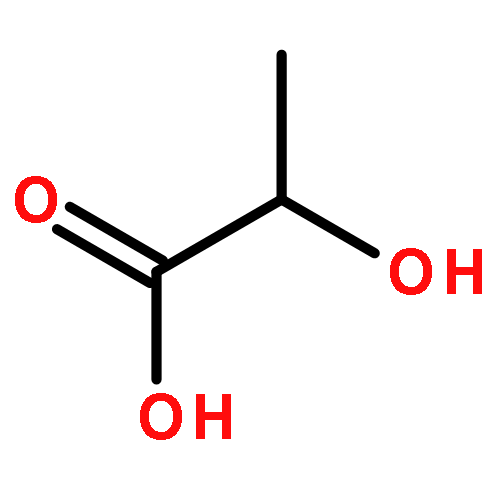
![Poly[imino[(1S)-1-(carboxymethyl)-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26100/26063-13-8.png)
![Poly[imino[(1S)-1-(carboxymethyl)-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26100/26063-13-8_b.png)


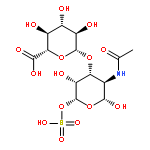
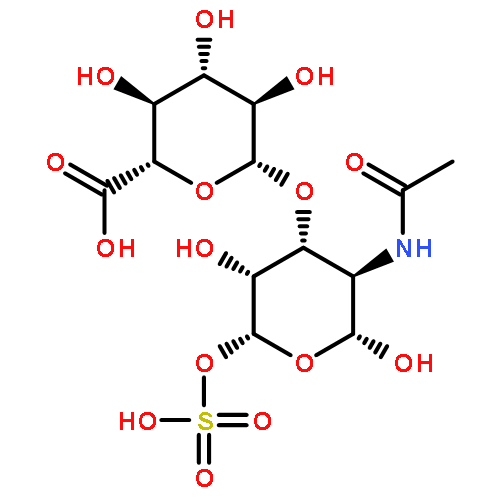




![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3_b.png)
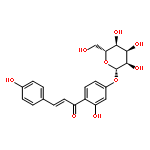



![(e)-1-(2,4-dihydroxyphenyl)-3-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]prop-2-en-1-one](http://img.cochemist.com/ccimg/5100/5041-81-6.png)
![(e)-1-(2,4-dihydroxyphenyl)-3-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]prop-2-en-1-one](http://img.cochemist.com/ccimg/5100/5041-81-6_b.png)


![Pregna-1,4-diene-3,20-dione,9-fluoro-11,17-dihydroxy-16-methyl-21-[(methylsulfonyl)oxy]-, (11b,16a)-](http://img.cochemist.com/ccimg/2300/2265-22-7.png)
![Pregna-1,4-diene-3,20-dione,9-fluoro-11,17-dihydroxy-16-methyl-21-[(methylsulfonyl)oxy]-, (11b,16a)-](http://img.cochemist.com/ccimg/2300/2265-22-7_b.png)




![(2s)-7-hydroxy-2-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]-2,3-dihydrochromen-4-one](http://img.cochemist.com/ccimg/600/551-15-5.png)
![(2s)-7-hydroxy-2-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]-2,3-dihydrochromen-4-one](http://img.cochemist.com/ccimg/600/551-15-5_b.png)







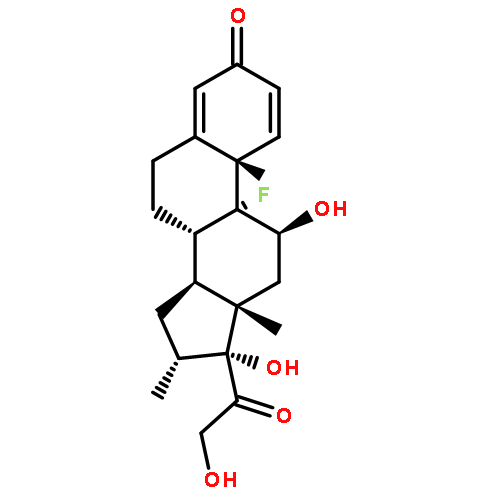






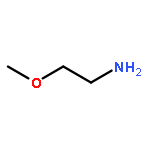
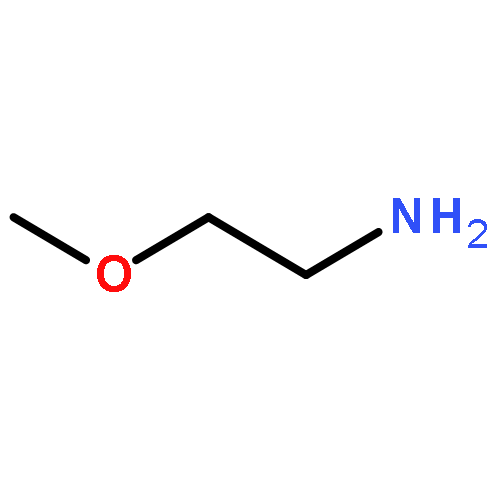


](http://img.cochemist.com/ccimg/27000/26913-06-4.png)
](http://img.cochemist.com/ccimg/27000/26913-06-4_b.png)




![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)