Co-reporter:Hailong Sun, Bifeng Zhu, Zhuang Wu, Xiaoqing Zeng, Helmut Beckers, and William S. Jenks
The Journal of Organic Chemistry 2015 Volume 80(Issue 3) pp:2006-2009
Publication Date(Web):January 13, 2015
DOI:10.1021/jo502821y
Transient carbonyl nitrenes RC(O)N, formed during thermal- or photoinduced decomposition of carbonyl azides RC(O)N3, are highly liable to the Curtius rearrangement, producing isocyanates RNCO in almost quantitative yield. Contrary to common belief, we found a thermally persistent triplet carbonyl nitrene, FC(O)N, that can be produced by flash pyrolysis of FC(O)N3 in 49% yield. The computed CBS-QB3 activation barrier for the thermal decomposition of FC(O)N3 to FC(O)N is 29 kJ mol–1 lower than that for a concerted pathway producing FNCO.
Co-reporter:Stacey A. Stoffregen, Stephanie Y. Lee, Pearl Dickerson and William S. Jenks
Photochemical & Photobiological Sciences 2014 vol. 13(Issue 2) pp:431-438
Publication Date(Web):06 Jan 2014
DOI:10.1039/C3PP50382H
CASSCF and multireference MP2 calculations were carried out on thiophene-S-oxide (TO) and selenophene-Se-oxide (SeO), comparing the energies of the ground state to the first two electronically excited singlet and triplet states, using constrained optimizations and multiple fixed S–O or Se–O distances. For both molecules, one of the two triplet states smoothly dissociates to yield O(3P) with little or no barrier. Single point calculations are consistent with the same phenomenon occurring for dibenzothiophene-S-oxide (DBTO). This provides an explanation for the inefficient unimolecular photochemical dissociation of O(3P) from DBTO despite a phosphorescence energy below that of S–O dissociation, i.e., that S–O scission probably occurs from a spectroscopically unobserved triplet (T2) state.
Co-reporter:Matthew P. Sherman and William S. Jenks
The Journal of Organic Chemistry 2014 Volume 79(Issue 19) pp:8977-8983
Publication Date(Web):September 8, 2014
DOI:10.1021/jo500664e
Computational methods are used to investigate the mechanism by which fluorination of acetylnitrene reduces the stabilization of the singlet configuration. ΔEST is made more positive (favoring the triplet state) by 1.9, 1.3, and 0.7 kcal/mol by the addition of the first, second, and third fluorine, respectively, at the CR-CC(2,3)/6-311(3df,2p)//B3LYP/6-31G(d,p) level of theory. Smaller effects observed with substitution of β-fluorines in propanoylnitrene derivatives and examination of molecular geometries and orbitals demonstrate that the effect is due to inductive electron withdrawal by the fluorines, rather than hyperconjugation.
Co-reporter:Timothy Hathway;Deborah Lipman Chernyshov
Journal of Physical Organic Chemistry 2011 Volume 24( Issue 12) pp:1151-1156
Publication Date(Web):
DOI:10.1002/poc.1839
Abstract
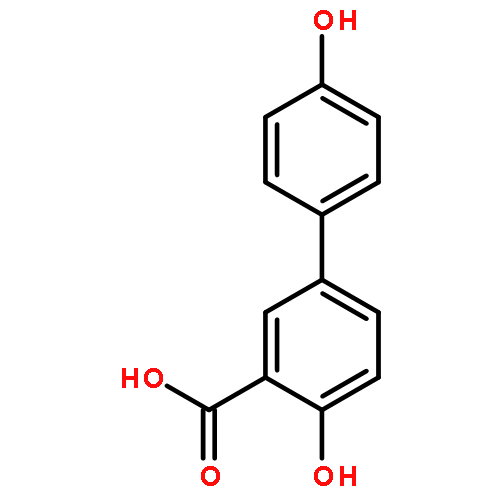
The selectivity of hydroxylation of the distal rings of 4-phenylbenzoic acid, 4-phenylsalicylic acid, and 5-phenylsalicylic acid were determined using partial TiO2-mediated photocatalytic degradation and photo-Fenton conditions. This separation of the binding site from the phenyl group being hydroxylated allows a less-biased evaluation. The hydroxylation regiochemistry behaves as qualitatively expected for an electrophilic reaction, given the assumption that 4-carboxyphenyl is a slightly electron-withdrawing substituent. Selectivity for hydroxylation of the distal phenyl in 4- and 5-phenylsalicylic acid is reversed, due to the reversal of the electronic demand, while adsorption to the TiO2 surface is assumed to be analogous for the two structures. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Erin M. Rockafellow, Jessica M. Haywood, Travis Witte, Robert S. Houk, and William S. Jenks
Langmuir 2010 Volume 26(Issue 24) pp:19052-19059
Publication Date(Web):November 12, 2010
DOI:10.1021/la1026569
This work describes the preparation of a selenium-modified TiO2 photocatalyst and a preliminary evaluation of its photocatalytic activity. Se-TiO2 displayed greater visible absorption than undoped TiO2 and was still capable of degrading quinoline at a slightly faster rate than undoped TiO2 under UV light. Se-TiO2 was also able to degrade organic molecules under purely visible light by a single electron transfer pathway. Irradiation with >435 nm light showed no evidence of efficient production of HO•-like species. Se-TiO2 was also examined under hypoxic conditions, where the Se atoms were capable of trapping photogenerated electrons as evidenced by XPS.
Co-reporter:Erin M. Rockafellow, Xiaowen Fang, Brian G. Trewyn, Klaus Schmidt-Rohr and William S. Jenks
Chemistry of Materials 2009 Volume 21(Issue 7) pp:1187
Publication Date(Web):March 16, 2009
DOI:10.1021/cm8019445
13C-modified TiO2 was prepared to facilitate study of the dopant atoms and trace their chemical fate throughout the process. In the preannealed material, NMR showed strong evidence of many Ti−O−C bonds. After annealing, surface-bound coke is a major component. NMR also showed that a washing step before annealing led to the generation of orthocarbonate (C(OR)4) centers, observed at 126 ppm, which are located deep inside the TiO2 particles. Both NMR and XPS confirmed the presence of small amounts of regular sp2-hybridized carbonate species in all briefly annealed samples, while annealing for longer times led to a reduction removal of the COn centers. Quantitative NMR also shows the degree of carbon loss that accompanies annealing. Some variation in the chemical degradation of quinoline is noted among the catalysts, but coke-containing TiO2 catalysts are not qualitatively better catalysts for use with visible light with this substrate.
Co-reporter:Timothy Hathway, Erin M. Rockafellow, Youn-Chul Oh, William S. Jenks
Journal of Photochemistry and Photobiology A: Chemistry 2009 Volume 207(2–3) pp:197-203
Publication Date(Web):25 September 2009
DOI:10.1016/j.jphotochem.2009.07.010
Tungsten-modified titanium dioxide catalysts prepared from sol–gel methods and obtained commercially were compared for their photocatalytic activity using mechanistic probes designed to examine chemical pathways of oxidation. No special visible absorbance was noted for the sol–gel catalysts. However, an increase in the single-electron transfer chemistry with the presence of WOx was noted, and a distinct wavelength dependence on the product ratios.
Co-reporter:Youn-Chul Oh, Yun Bao, William S Jenks
Journal of Photochemistry and Photobiology A: Chemistry 2003 Volume 161(Issue 1) pp:69-77
Publication Date(Web):17 November 2003
DOI:10.1016/S1010-6030(03)00272-7
The initial step of TiO2-mediated photocatalytic degradation of dimethyl phenylphosphonate (DMPP), labeled with or deuteria in the methoxy groups, results in products due to ring hydroxylation and demethylation. The labeling experiments clearly demonstrate that the methyl group is lost, rather than a methoxy group, resulting in a labeled phosphonic mono-acid. Results from deuterium isotope experiments are more ambiguous.
Co-reporter:Ryan D. McCulla;Jerry W. Cubbage
Journal of Physical Organic Chemistry 2002 Volume 15(Issue 2) pp:71-77
Publication Date(Web):5 DEC 2001
DOI:10.1002/poc.464
The barriers to pyrolytic unimolecular elimination by ethyl methanesulfonate and ethyl methanesulfinate were calculated for both five-membered ring eliminations that yield acetaldehyde and six-membered ring eliminations that yield ethylene. The experimental observation that related sulfinates undergo the five-centered elimination and sulfonates the six-centered elimination is reproduced by the calculations and rationalized in terms of a nucleophilicity/electrophilicity matching issue in the former and a charge-separated transition state in the latter. Copyright © 2001 John Wiley & Sons, Ltd.
Co-reporter:Xiaojing Li, Jerry W Cubbage, William S Jenks
Journal of Photochemistry and Photobiology A: Chemistry 2001 Volume 143(Issue 1) pp:69-85
Publication Date(Web):1 October 2001
DOI:10.1016/S1010-6030(01)00472-5
The early degradation product distributions from TiO2-mediated photocatalytic degradations of a series of multiply hydroxylated benzenes and their methoxylated analogs is reported. The methoxylated compounds show a distinct trend away from ring-opening reactions that are attributed to electron transfer chemistry towards hydroxylations and demethylations that are attributed to hydroxyl-type chemistry. The initial rates of the reactions suggest that this is not simply due to the availability of the extra carbon sites. It is instead hypothesized that the compounds with hydroxyl groups are bound to the TiO2 in such a way as to facilitate electron transfer and its subsequent chemistry. Addition of isopropyl alcohol to the degradations slows the degradations of the methoxylated compounds more than it does the hydroxylated compounds and particularly inhibits the demethylation and hydroxylation reactions. It is suggested that the question of whether an organic substrate must be bound to the photocatalyst at the time of excitation may be dependent on the type of chemistry being observed.
Co-reporter:Erin M. Rockafellow, Laine K. Stewart, William S. Jenks
Applied Catalysis B: Environmental (7 September 2009) Volume 91(Issues 1–2) pp:554-562
Publication Date(Web):7 September 2009
DOI:10.1016/j.apcatb.2009.06.027
.jpg)






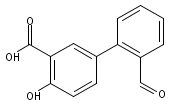
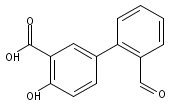
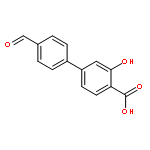
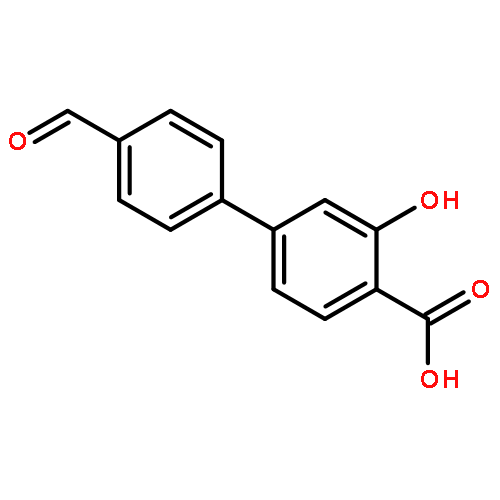
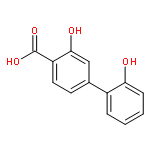
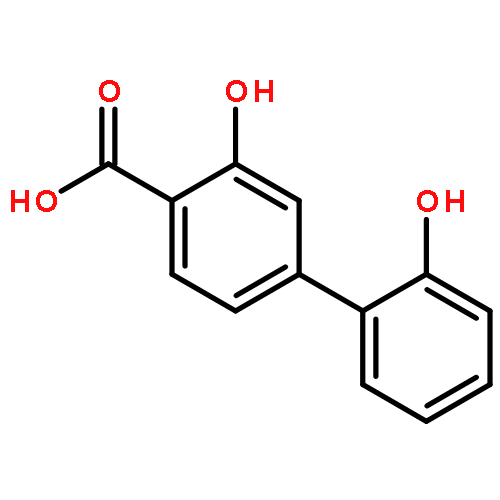
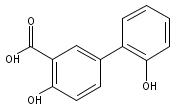
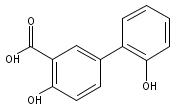
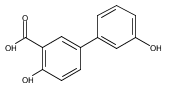
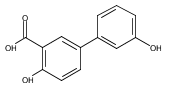
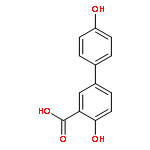
![3-Hydroxy-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/4500/4482-27-3.png)
![3-Hydroxy-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/4500/4482-27-3_b.png)





